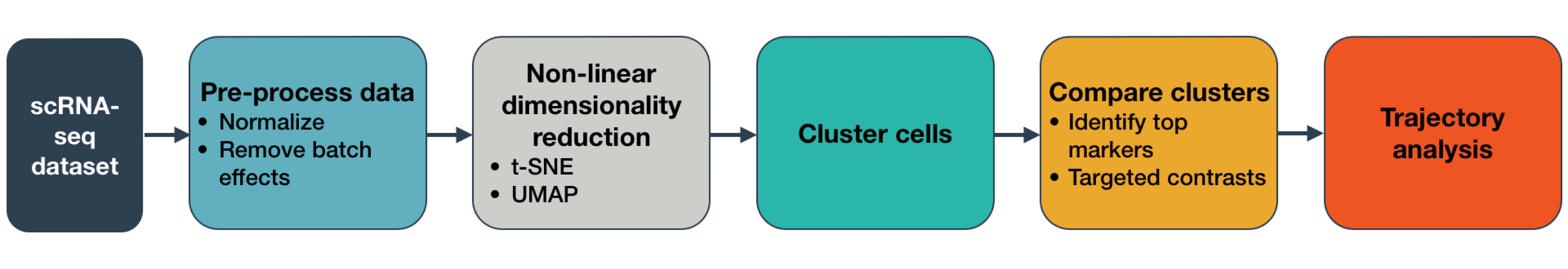
In this tutorial we will perform trajectory analysis using monocle3. You can find out more about the theory behind trajectory analysis in our slide deck. We have already analysed the trajectory of our sample using the ScanPy toolkit in another tutorial: Inferring Trajectories using Scanpy. However, trajectory analysis is quite sensitive and some methods work better for specific datasets. In this tutorial, you will perform the same steps but using a different method for inferring trajectories. You will then compare the results, usability and outcomes! Sounds exciting, let’s dive into that!
Tools are frequently updated to new versions. Your Galaxy may have multiple versions of the same tool available. By default, you will be shown the latest version of the tool. This may NOT be the same tool used in the tutorial you are accessing. Furthermore, if you use a newer tool in one step, and try using an older tool in the next step… this may fail! To ensure you use the same tool versions of a given tutorial, use the Tutorial mode feature.
Open your Galaxy server
Click on the curriculum icon on the top menu, this will open the GTN inside Galaxy.
Navigate to your tutorial
Tool names in tutorials will be blue buttons that open the correct tool for you
Note: this does not work for all tutorials (yet)
You can click anywhere in the grey-ed out area outside of the tutorial box to return back to the Galaxy analytical interface
Warning: Not all browsers work!
We’ve had some issues with Tutorial mode on Safari for Mac users.
Try a different browser if you aren’t seeing the button.
In the previous tutorials, we first created an AnnData object and performed downstream analysis on that file. However, Monocle3 uses another datatype which is Cell Data Set (CDS). To be able to infer trajectories in Monocle, we need to transform our AnnData object into CDS file. And guess what - we already have a tutorial for that! We did it in format conversion tutorial, in the Anndata -> Cell Data Set (CDS) subsection. To better understand the structure of CDS object and learn how to create it from expression matrix, cell and gene annotations, it is highly recommended that you complete the mentioned tutorial before importing the prepared CDS file.
You have two options for uploading the dataset. Importing via history is often faster.
Hands On: Option 1: Data upload - Import history
You can import history where we went from AnnData to CDS file. Then you will also have access to extracted cell metadata, gene metadata, and an expression matrix. If you want to get the CDS file alone, you can get it in this input history.
Open the link to the shared history
Click on the Import this history button on the top left
Enter a title for the new history
Click on Copy History
Renamegalaxy-pencil the the history to your name of choice.
Click galaxy-uploadUpload at the top of the activity panel
Select galaxy-wf-editPaste/Fetch Data
Paste the link(s) into the text field
Press Start
Close the window
Monocle3 turns the expression matrix, cell and gene annotations into an object called cell_data_set (CDS), which holds single-cell expression data. We provided you with the CDS file already, but it was created by combining the three mentioned elements. Check out data conversion tutorial to see how to do it!
Here is what Monocle3 documentation says about the three input files required to create a CDS object:
expression_matrix: a numeric matrix of expression values, where rows are genes, and columns are cells. Must have the same number of columns as the cell_metadata has rows and the same number of rows as the gene_metadata has rows.
cell_metadata: a data frame, where rows are cells, and columns are cell attributes (such as cell type, culture condition, day captured, etc.)
gene_metadata: a data frame, where rows are features (e.g. genes), and columns are gene attributes, such as biotype, gc content, etc. One of its columns should be named “gene_short_name”, which represents the gene symbol or simple name (generally used for plotting) for each gene.
Once you get the CDS file in your history, let’s have a closer look at this dataset to understand it a little bit better.
Question
What are the dimensions of the created CDS object?
Just click on that dataset - on the preview you’ll see that the dimensions are 15395 x 8569 - so exactly as we predicted genes x cells!
Figure 2: Workflow provided by Monocle3 documentation
We will follow those steps and see how it all works in practice.
Pre-processing
In Galaxy, there are currently 2 methods of initial dimensionality reduction included in the pre-processing step: principal component analysis (PCA) and latent semantic indexing (LSI).
Given that PCA is more commonly used, and it allows us to perform further steps on the CDS object, we’ll use this method. There is one parameter here that has a great impact on how our analysis will look - the dimensionality of the initially reduced space. This is a highly subjective choice - you will want to test a lot of different parameters on your dataset. After much trial and error, we were able to find the value that made the most sense biologically. Have a look at the image below to see how different values affect the outcomes.
Figure 3: Different outputs depending on the number of dimensions that the space was reduced to during pre-processing. Currently the label size in Monocle graphs in Galaxy is a nightmare, so for clarity additional legend was added to this image. We're working on the label size so that you can generate clear, readable and pretty graphs. However in this particular figure, the main point is to show the differences in shapes depending on the num-dim.
Question
Looking at the image above, which value would you choose?
It might be hard to tell at this point without any explanation! Don’t worry, after a few more steps you’ll understand what all those colors mean and how to generate those plots. But look - we want to infer trajectories and find relationships between cells, ideally we would see the development of cells or transition from one type to another. In the graphs where num-dim is from 10 to 200, you see that the clusters are quite disjoint, while we want to see smooth transitions from one to another. I can tell you now that we’ll go ahead with the value of 250. We’re not choosing 300, because the arrangement of the cells on that graph is not really biologically relevant.
Hands On: Pre-processing
Monocle3 preprocess ( Galaxy version 0.1.4+galaxy0) with the following parameters:
param-file“Input object in RDS format”: output of Monocle3 createtool
“The dimensionality of the reduced space.”: 250
Dimensionality reduction
Now it’s time for the proper dimensionality reduction, to turn the original thousands of dimensions (genes), into a 2-dimensional graph. There are several algorithms to do this: UMAP, tSNE, PCA and LSI (only possible when preprocess_method is set to LSI), but due to the same reasons as above, we’ll use UMAP (most common + allows further operations + best results). But I’ll let you see how the outputs from the other algorithms look to convince you that UMAP is indeed the best for this dataset. Of course, it’s possible that by choosing different pre-processing values, tSNE or PCA plots would look better, so don’t be afraid to play around with the parameters and test them!
Figure 4: The preview of alignment of cell types depending on the algorithm of dimensionality reduction that was chosen: UMAP, tSNE, PCA, LSI. The methods were applied to the output of the PCA-preprocessed data (except LSI method which was called on LSI-preprocessed data). LSI failed in forming distinct cell groups, PCA managed to cluster cells according to their types but tSNE did it more precisely. However, UMAP gave the best results, not only showing distinct cell type groups, but also ordering them in a way that makes sense biologically - DN to T-mat.
Hands On: Dimensionality reduction
Monocle3 reduceDim ( Galaxy version 0.1.4+galaxy0) with the following parameters:
param-file“Input object in RDS format”: output of Monocle3 preprocesstool
Plotting
Alright, now let’s have a look at our output! Above you got a sneak peek of how the plot would look, but now you’ll generate the plots on your own!
Thanks to the fact that we provided Monocle3 with annotated data, we can now color the cells by any attribute that was in the cell metadata file! Similarly to the previous tutorial, we’ll color them by cell type, genotype, batch and sex. At least for now…
Hands On: Plotting
Monocle3 plotCells ( Galaxy version 0.1.5+galaxy1) with the following parameters:
param-file“Input object in RDS format”: output of Monocle3 reduceDimtool
“The cell attribute (e.g. the column of pData(cds)) to map to each cell’s color, or one of {cluster, partition, pseudotime}.”: cell_type
“If set, display the cell group names directly on the plot. Otherwise include a color legend on the side of the plot.”: param-toggleNo
Rename galaxy-pencil the output: Cell type plot
Monocle3 plotCells ( Galaxy version 0.1.5+galaxy1) with the following parameters:
param-file“Input object in RDS format”: output of Monocle3 reduceDimtool
“The cell attribute (e.g. the column of pData(cds)) to map to each cell’s color, or one of {cluster, partition, pseudotime}.”: genotype
“If set, display the cell group names directly on the plot. Otherwise include a color legend on the side of the plot.”: param-toggleNo
Rename galaxy-pencil the output: Genotype plot
Monocle3 plotCells ( Galaxy version 0.1.5+galaxy1) with the following parameters:
param-file“Input object in RDS format”: output of Monocle3 reduceDimtool
“The cell attribute (e.g. the column of pData(cds)) to map to each cell’s color, or one of {cluster, partition, pseudotime}.”: batch
“If set, display the cell group names directly on the plot. Otherwise include a color legend on the side of the plot.”: param-toggleNo
Rename galaxy-pencil the output: Batch plot
Monocle3 plotCells ( Galaxy version 0.1.5+galaxy1) with the following parameters:
param-file“Input object in RDS format”: output of Monocle3 reduceDimtool
“The cell attribute (e.g. the column of pData(cds)) to map to each cell’s color, or one of {cluster, partition, pseudotime}.”: sex
“If set, display the cell group names directly on the plot. Otherwise include a color legend on the side of the plot.”: param-toggleNo
Rename galaxy-pencil the output: Sex plot
The Previous tutorial discussed in detail the biological interpretation of data, so we will quickly go through similar analysis to see if the results are consistent and if we can draw any new conclusions.
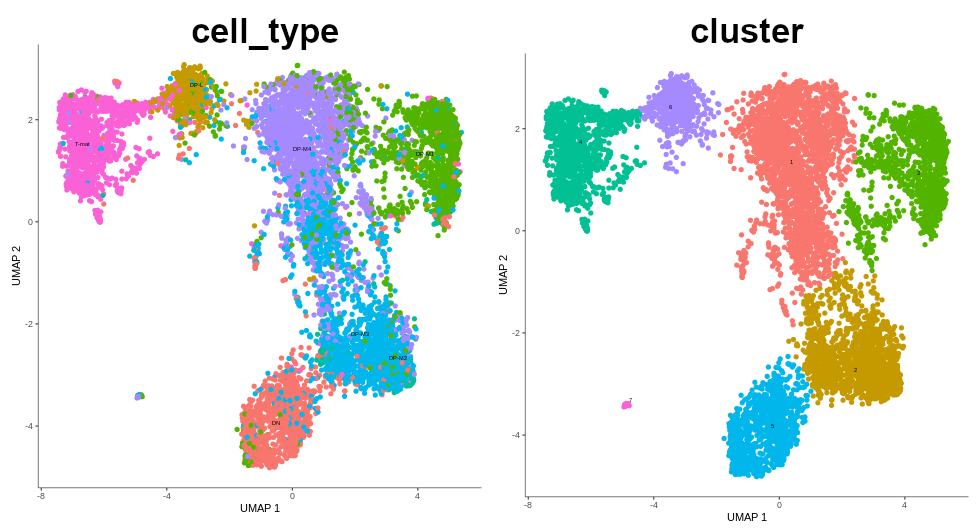
As a reminder, here’s the comparision between cell type annotation done in the other tutorial using Scanpy, and the output from the previous step of this Monocle tutorial. The main difference is that Scanpy was used to identify the cell types and assign them to clusters. That data was then passed on to Force-Directed + PAGA algorithms to infer trajectory, and then the arrangement of the cell groups changed a bit. In Monocle, trajectory analysis will be based on the clustering you see now.
Figure 5: Cells coloured in Monocle by their type.
In the mentioned tutorial, we annotated the cells so that we know what type they are. Above you can see how Monocle used this information to colour cells by their type. It is important to see the ‘form’ of the graph and how the cell types are arranged within its confines. But why is it important? Well, in Monocle, our trajectory analysis will be based on the same arrangement of the cells that you see now. And if you now recall our choice of the number of dimensions during pre-processing, you’ll understand why it was crucial - choosing the right value at the beginning determines the ‘shape’ of the graph that is then retained for the trajectory analysis. Therefore, the fact that on the plot above we clearly see DN cells on one side of the graph and T-mat on the other, going through DP cells, looks promising. But there is DP-M1 group that suspiciously branches out… Let’s investigate that and wait until the trajectory is inferred!
Question: Genotype
Based on our results, can we confirm findings from the previous tutorial that DP-L and mature T-cells (particularly the top half) are missing some knockout cells?
Indeed, that’s what we see in our graph! But look closer, there is something more…Additionally, we also discovered that the vast majority of DP-M1 is only wildtype cells. That’s interesting, isn’t it?
Question: Batch effect
Can we confirm the previous findings that DP-L looks to be mainly comprised of N705?
Both DP-L and DP-M1 seem to consist mostly of N705 and N706. There might be indeed a bit of batch effect, so you could consider using batch correction on this dataset. In the absence of batch correction, we will focus on those clusters where there is batch mixing for biological interpretation. Finally, we will look at the confounding effect of sex.
There are also no female cells in DP-L and DP-M1. The one female sample - which is one of the mere three knockout samples - seems to be distributed in the same areas as the knockout samples at large. Luckily, this doesn’t seem to be a confounding factor and we can still learn from our data.
Clustering
Don’t get confused - we haven’t clustered our cells yet, for now we have only plotted them based on cell type annotation. Now it’s time to create clusters, which - in an ideal world where all computation picks up the exact biological phenomenons - would yield the same areas as the clusters determined by the Scanpy algorithms. Is this the case here? Do Monocle and Scanpy identify the same clusters?
Monocle uses a technique called “community detection” (Traag et al. 2019) to group cells. This approach was introduced by Levine et al. 2015 as part of the phenoGraph algorithm.
Monocle also divides the cells into larger, more well separated groups called partitions, using a statistical test from Wolf et al. 2019, introduced as part of their PAGA algorithm.
Clusters are particularly useful while trying to assign cells to a certain type, because they are based on the similarity in gene expression. The relationships between different clusters are analysed to identify possible trajectories.
Partitions, meanwhile, are larger groups of cells that usually contain several clusters. Trajectory inference is performed only within one partition, so it is essential that all the cells that we want to analyse in pseudotime belong to the same partition.
Hands On: Clustering
Monocle3 partition ( Galaxy version 0.1.4+galaxy0) with the following parameters:
param-file“Input object in RDS format”: output of Monocle3 reduceDimtool
“Resolution of clustering result, specifying the granularity of clusters. Not used by default and the standard igraph louvain clustering algorithm will be used.”: 0.00015
“The q-value threshold used to determine the partition of cells.”: 1.0
The clusters and partitions are now stored in your CDS file. To see them, just plot the output, coloring the cells with the corresponding attributes.
Monocle3 plotCells ( Galaxy version 0.1.5+galaxy1) with the following parameters:
param-file“Input object in RDS format”: output of Monocle3 partitiontool
“The cell attribute (e.g. the column of pData(cds)) to map to each cell’s color, or one of {cluster, partition, pseudotime}.”: partition
Rename galaxy-pencil the output: Partition plot
Monocle3 plotCells ( Galaxy version 0.1.5+galaxy1) with the following parameters:
param-file“Input object in RDS format”: output of Monocle3 partitiontool
“The cell attribute (e.g. the column of pData(cds)) to map to each cell’s color, or one of {cluster, partition, pseudotime}.”: cluster
Rename galaxy-pencil the output: Cluster plot
Sometimes it might happen that cells are grouped into several partitions, while you want them all to be in just one in order to perform trajectory analysis on all of them. Then, you can try to increase the q-value threshold that is used to determine the partition of cells.
Figure 9: q-value threshold affecting the span of partition. Note that 0.05 is the default value.
When using standard igraph louvain clustering, the value of resolution parameter is by default set to NULL, which means that it is determined automatically. If you are not satisfied with the results of the standard igraph louvain clustering, you may set the resolution value manually, and thus specify the granularity of the clustering.
Figure 10: Different granularity of clusters based on the resolution set automatically and manually.
Warning: Ambiguous clusters!
As mentioned above, standard igraph louvain clustering determines the resolution automatically, unless the specific value is provided by the user. Therefore, it sometimes returns slightly different outputs. To ensure that your clusters are reproducible, you might want to pass a certain value to the resolution parameter. In case of our data, the resolution value of 0.00015 gave the same results as the best output of igraph louvain clustering, and ensured reproducibility.
Figure 11: Standard igraph louvain clustering giving different results despite the same input becasue of the automatic determination of resolution vaule.
If we compare the annotated cell types and the clusters that were just formed, we see that they nicely correspond to one another.
Figure 12: Comparision between annotated cell types and formed clusters.
Gene expression
We haven’t looked at gene expression yet! This step is particularly important when working with data which is not annotated. Then, based on the expression of marker genes, you are able to identify which clusters correspond to which cell types. This is indeed what we did in the previous tutorial using scanpy. We can do the same using Monocle3! Since we work on annotated data, we can directly check if the expressed genes actually correspond to the previously assigned cell types. If they do, that’s great - if two different methods are consistent, that gives us more confidence that our results are valid.
Below is the table that we used in the previous tutorial to identify the cell types.
Marker
Cell type
Il2ra
Double negative (early T-cell)
Cd8b1, Cd8a, Cd4
Double positive (middle T-cell)
Cd8b1, Cd8a, Cd4 - high
Double positive (late middle T-cell)
Itm2a
Mature T-cell
Aif1
Macrophages
Hba-a1
RBC
Hands On: Gene expression
Monocle3 plotCells ( Galaxy version 0.1.5+galaxy1) with the following parameters:
param-file“Input object in RDS format”: output of Monocle3 partitiontool
“The cell attribute (e.g. the column of pData(cds)) to map to each cell’s color, or one of {cluster, partition, pseudotime}.”: cell_type
“A list of gene IDs/short names to plot.”: Il2ra,Cd8b1,Cd8a,Cd4,Itm2a,Aif1,Hba-a1
Figure 13: Expression of the genes across analysed sample
Question: Genes and cell types
Based on the gene expression graph that we just generated, the table above and your knowledge from the previous tutorial, how would you interpret the results?
Il2ra: expressed in the cluster where DN cells are - an indication of where the trajectory should start
Cd8b1, Cd8a: expressed in the areas where middle DP were assigned - great
high Cd4: mostly in late DP cluster - as expected
Itm2a: expressed in mature T-cells - tells us where the trajectory should end
Aif1: nothing here - correct! We filtered out macrophages from the sample
Hba-a1: look at this! Very high expression in just one, tiny bit, which hasn’t even been grouped into a cluster! So wait, why do we see hemoglobin gene here? Do we have red blood cells in our sample?! Let’s investigate that further…
The Hba-a1 gene creates hemoglobin which is found in red blood cells. This is highly expressed in a tiny bit of the middle DP cluster. Interestingly, it forms a a clearly visible, distinct, little branch. Hemoglobin should NOT be found in T-cells. However, if you remember, the gene was found to be expressed in the previous Scanpy tutorial (see the image below). That marker appeared throughout the entire sample in low numbers, suggesting some background contamination of red blood cell debris in the cell samples during library generation. Unlike Scanpy, Monocle algorithms allowed us to gather the cells expressing that gene into a distinct group! That’s great!
Figure 14: Hemoglobin across clusters - comparision between Monocle and Scanpy
Top marker genes
Here we used a priori knowledge regarding the marker genes. If we wanted to approach this problem in an unsupervised manner, we could use Monocle to tell us the top marker genes in each group of cells. This is very useful if we are trying to identify a cell type, or if we want to find novel marker genes for known cell types.
Question
If I cluster cells that are not annotated, can I assign clusters to a cell type based on gene expression using Monocle3?
Theoretically…yes! That’s the point of the clustering and gene expression analysis. However, as of the writing of this tutorial, this function hasn’t been turned into a Galaxy tool yet and is only available in R.
Hands On: Top marker genes
Monocle3 top markers ( Galaxy version 0.1.5+galaxy0) with the following parameters:
param-file“Input Object”: output of Monocle3 partitiontool
“Group cell by”: cell_type
Rename galaxy-pencil the tabular output: Top markers table
Rename galaxy-pencil the pdf output: Top markers plot
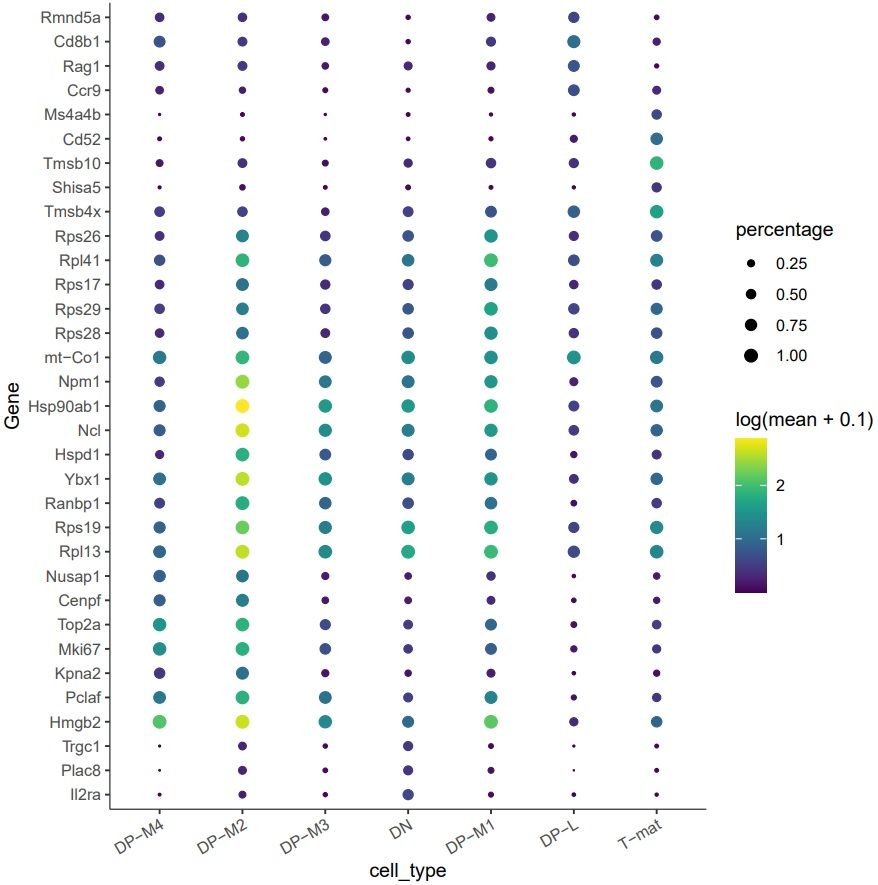
Figure 15: Identification of the genes most specifically expressed in groups of cells.
Question
What genes are uniquely expressed in DP-M1?
By looking at the table, you might give the 5 top gene IDs expressed in DP-M1. To save you some time and make the analysis more readable, we converted the gene IDs to gene names and they are as follows: Rps17, Rpl41, Rps26, Rps29, Rps28. They are all ribosomal! [You can do this yourself if you want by following this section of a previous tutorial that uses the gene names in one object to add to a table of Ensembl IDs. These ribosomal differences might be due to housekeeping background, cell cycling, or even something more bioligically interesting…or all three!
The plot also indicates other specifically expressed genes, such as Hmgb2, Pclaf, Rpl13, Rps19, Ybx1, Ncl, Hsp90ab1, Npm1.
Whenever you want to explore what might be the function of a particular cluster or why it branches out from the trajectory, check the top markers for that cluster to draw biological conclusions. Thank you Maths!
But what if you want to know how gene expression changes across a trajectory? This is where Monocle is particularly powerful. But in order to do that, we have to infer that trajectory first!
Learn the trajectory graph
We’re getting closer and closer! The next step is to learn the trajectory graph, which means to fit a principal graph within each partition. In that way, we’ll ‘connect’ the existing clusters by creating a path between them.
Hands On: Learn graph
Monocle3 learnGraph ( Galaxy version 0.1.4+galaxy0) with the following parameters:
param-file“Input object in RDS format”: output of Monocle3 partitiontool
Again, the graph is now stored in your CDS file. To see it, just plot the output, you can color the cells by any attribute that you want. We’ll use cell types to see how they are connected.
Monocle3 plotCells ( Galaxy version 0.1.5+galaxy1) with the following parameters:
param-file“Input object in RDS format”: output of Monocle3 learnGraphtool
“The cell attribute (e.g. the column of pData(cds)) to map to each cell’s color, or one of {cluster, partition, pseudotime}.”: cell_type
As you can see, the learned trajectory path is just a line connecting the clusters. However, there are some important points to understand here.
If the resolution of the clusters is high, then the trajectory path will be very meticulous, strongly branched and curved. There’s a danger here that we might start seeing things that don’t really exist.
You can set an option to learn a single tree structure for all the partitions or use the partitions calculated when clustering and identify disjoint graphs in each. To make the right decision, you have to understand how/if the partitions are related and what would make more biolgical sense. In our case, we were only interested in a big partition containing all the cells and we ignored the small ‘dot’ classified as another partition.
There are many trajectory patterns: linear, cycle, bifurcation, tree and so on. Those patterns might correspond to various biological processes: transition events for different phases, cell cycle, cell differentiation. Therefore, branching points are quite important on the trajectory path. You can always plot them, param-toggle checking the correct box in Monocle3 plotCells.
As a reminder, here’s the comparision between our trajectory and the one from the previous tutorial, where we used Scanpy for clustering, and then appplied Force-Directed + PAGA algorithms to infer trajectory. As you remember from those tutorials, the arrangement of the cell groups changed a bit during these steps. Indeed - by using the mentioned methods, we get different graphs for clusters and trajectory, while in Monocle the general ‘shape’ of the graph stays the same from the beginning. ‘Leranig the trajectory’ step in Monocle is about finding a path, along which the cells can be then ordered in pseudotime.
Figure 17: Comparison between the trajectory inferred in the previous 'case study' tutorials (Scanpy + Force-Directed + PAGA) and the trajectory obtained in Monocle.
Pseudotime analysis
Finally, it’s time to see our cells in pseudotime! We have already learned a trajectory, now we only have to order the cells along it. Monocle3 requires information on where to start ordering the cells, so we need to provide it with this information. We annotated early T-cells as double negative (DN), so those will be our root cells!
To infer trajectories, we need data from cells at different points along a path of differentiation. The assumption is that in any given sample, some cells are further along a trajectory than others. This inferred temporal dimension is known as pseudotime. Pseudotime measures the cells’ progress through the transition. See Reid and Wernisch 2016 for more.
Hands On: Ordering the cells along trajectory
Monocle3 orderCells ( Galaxy version 0.1.4+galaxy0) with the following parameters:
param-file“Input object in RDS format”: output of Monocle3 learnGraphtool
“The cell phenotype (column in pdata) used to identify root principal nodes.”: cell_type
“The value in the cell phenotype column used to extract root nodes.”: DN
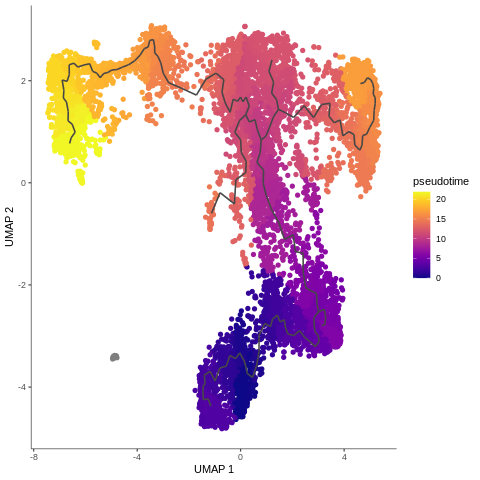
Alright - we were waiting for this plot the whole tutorial: once we have the cells ordered, we can finally color them by pseudotime!
Monocle3 plotCells ( Galaxy version 0.1.5+galaxy1) with the following parameters:
param-file“Input object in RDS format”: output of Monocle3 orderCellstool
Rename galaxy-pencil the output: Pseudotime plot
The method to specify the root cells shown above is not the only one available in Galaxy! However, it is probably the most intuitive one.
Annotated cell type as root cells
fill (–cell-phenotype) with the colname (heading) where the cell types are stored
fill (–root-type) with the name of the cell type that you want to start ordering from
Cell ID as root cell
fill (–root-cells) with the cell ID that you want to start ordering from
Now we can see how all our hard work has come together to give a final pseudotime trajectory analysis. DN cells gently switching to DP-M which change into DP-L to finally become mature T-cells. Isn’t it beautiful? But wait, don’t be too enthusiastic - why on earth DP-M1 group branches out? We didn’t expect that… What could that mean?
There are a lot of such questions in bioinformatics, and we’re always get excited to try to answer them. However, with analysing scRNA-seq data, it’s almost like you need to know about 75% of your data to make sure that your analysis is reasonable, before you can identify the 25% new information. Additionally, pseudotime analysis crucially depends on choosing the right analysis and parameter values, as we showed for example with initial dimensionality reduction during pre-processing. The outputs here, at least in our hands, are more sensitive to parameter choice than standard clustering analysis with Scanpy.
Last but not least, you can now identify genes that define the inferred trajectories.
Hands On: Differentially expressed genes
Monocle3 diffExp ( Galaxy version 0.1.4+galaxy1) with the following parameters:
param-file“Input object in RDS format”: output of Monocle3 orderCellstool
Rename galaxy-pencil the output: Differential gene expression table
Conclusion
congratulations Well done, you’ve made it to the end! You might want to consult your results with this control history (which also includes AnnData to CDS conversion), and check out the workflow for this tutorial. There is also a separate workflow for preparing the input files for Monocle3, starting from AnnData, and full analysis workflow. You can use them to accelerate analysis of your own data, paying attention to the requirements of the input data that are mentioned in this tutorial.
Figure 19: Full workflow for this tutorial, including AnnData to CDS format conversion.
If you’re following the Case Study tutorials from the beginning, you have already experienced what it’s like to analyse and question a dataset, potentially without clear cut-offs or clear answers. You now know that trajectory analysis is even more sensitive to parameter values, so it’s often trying to find the best set of values that would give the most reasonable results and go in accordance with biology. Moreover, not all trajectory analysis methods are designed to infer all kinds of biological processes - due to the fact that they use different algorithms, some would work better for analysing your sample. Since Monocle is quite widely used for trajectory analysis, it might be a good practice to compare its results with other methods. The more evidence you have to confirm your findings, the more confident you can be about their reliability!
We also post new tutorials / workflows there from time to time, as well as any other news.
point-right If you’d like to contribute ideas, requests or feedback as part of the wider community building single-cell and spatial resources within Galaxy, you can also join our Single cell & sPatial Omics Community of Practice.
Please also consider filling out the Feedback Form as well!
Key points
You should understand your data object sufficiently to be able to extract relevant information for further analysis.
Trajectory analysis is highly dependent on the parameter values you choose, as such ‘inferred relationships’ are a bigger mathematical leap. Therefore, you should always check if the output makes biological sense before proceeding to the next step.
Comparing the output of two different methods applied on the same dataset might be useful to confirm the results, to ensure that the findings are reliable and even sometimes to find a new piece of information.
Frequently Asked Questions
Have questions about this tutorial? Have a look at the available FAQ pages and support channels
Further information, including links to documentation and original publications, regarding the tools, analysis techniques and the interpretation of results described in this tutorial can be found here.
References
Levine, J. H., E. F. Simonds, S. C. Bendall, K. L. Davis, E.-ad D. Amir et al., 2015 Data-Driven Phenotypic Dissection of AML Reveals Progenitor-like Cells that Correlate with Prognosis. Cell 162: 184–197. 10.1016/j.cell.2015.05.047
Reid, J. E., and L. Wernisch, 2016 Pseudotime estimation: deconfounding single cell time series. Bioinformatics 32: 2973–2980. 10.1093/bioinformatics/btw372
Bacon, W. A., R. S. Hamilton, Z. Yu, J. Kieckbusch, D. Hawkes et al., 2018 Single-Cell Analysis Identifies Thymic Maturation Delay in Growth-Restricted Neonatal Mice. Frontiers in Immunology 9: 10.3389/fimmu.2018.02523
Traag, V. A., L. Waltman, and N. J. van Eck, 2019 From Louvain to Leiden: guaranteeing well-connected communities. Sci Rep 9: 10.1038/s41598-019-41695-z
Wolf, F. A., F. K. Hamey, M. Plass, J. Solana, J. S. Dahlin et al., 2019 PAGA: graph abstraction reconciles clustering with trajectory inference through a topology preserving map of single cells. Genome Biol 20: 10.1186/s13059-019-1663-x
Feedback
Did you use this material as an instructor? Feel free to give us feedback on how it went.
Did you use this material as a learner or student? Click the form below to leave feedback.
Hiltemann, Saskia, Rasche, Helena et al., 2023 Galaxy Training: A Powerful Framework for Teaching! PLOS Computational Biology 10.1371/journal.pcbi.1010752
Batut et al., 2018 Community-Driven Data Analysis Training for Biology Cell Systems 10.1016/j.cels.2018.05.012
@misc{single-cell-scrna-case_monocle3-trajectories,
author = "Julia Jakiela",
title = "Inferring single cell trajectories with Monocle3 (Galaxy Training Materials)",
year = "",
month = "",
day = "",
url = "\url{https://training.galaxyproject.org/training-material/topics/single-cell/tutorials/scrna-case_monocle3-trajectories/tutorial.html}",
note = "[Online; accessed TODAY]"
}
@article{Hiltemann_2023,
doi = {10.1371/journal.pcbi.1010752},
url = {https://doi.org/10.1371%2Fjournal.pcbi.1010752},
year = 2023,
month = {jan},
publisher = {Public Library of Science ({PLoS})},
volume = {19},
number = {1},
pages = {e1010752},
author = {Saskia Hiltemann and Helena Rasche and Simon Gladman and Hans-Rudolf Hotz and Delphine Larivi{\`{e}}re and Daniel Blankenberg and Pratik D. Jagtap and Thomas Wollmann and Anthony Bretaudeau and Nadia Gou{\'{e}} and Timothy J. Griffin and Coline Royaux and Yvan Le Bras and Subina Mehta and Anna Syme and Frederik Coppens and Bert Droesbeke and Nicola Soranzo and Wendi Bacon and Fotis Psomopoulos and Crist{\'{o}}bal Gallardo-Alba and John Davis and Melanie Christine Föll and Matthias Fahrner and Maria A. Doyle and Beatriz Serrano-Solano and Anne Claire Fouilloux and Peter van Heusden and Wolfgang Maier and Dave Clements and Florian Heyl and Björn Grüning and B{\'{e}}r{\'{e}}nice Batut and},
editor = {Francis Ouellette},
title = {Galaxy Training: A powerful framework for teaching!},
journal = {PLoS Comput Biol}
}
Funding
These individuals or organisations provided funding support for the development of this resource
Questions:

Open image in new tab
 Open image in new tab
Open image in new tab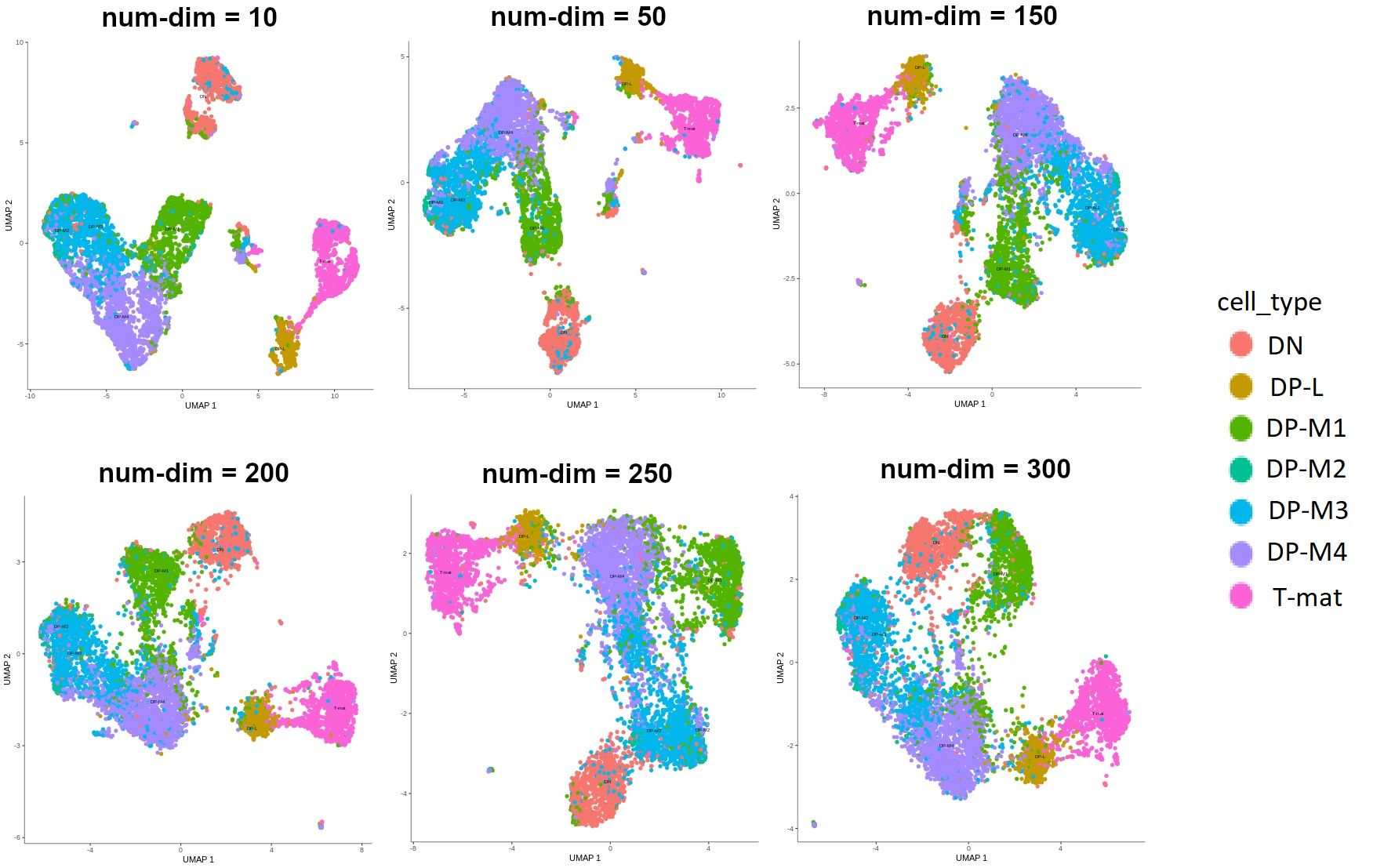 Open image in new tab
Open image in new tab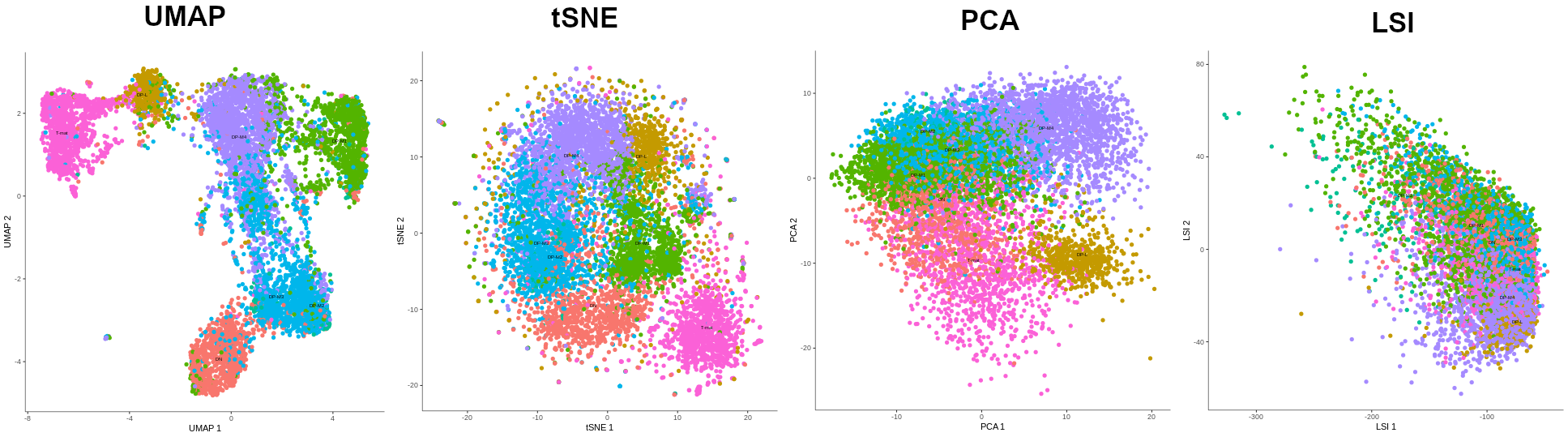 Open image in new tab
Open image in new tab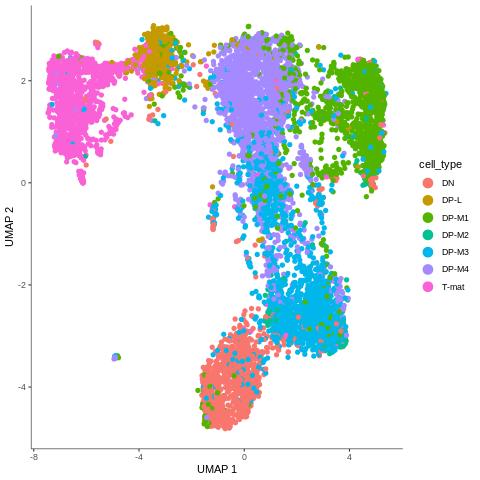 Open image in new tab
Open image in new tabOpen image in new tab
Open image in new tab
Open image in new tab
Open image in new tab
Open image in new tab
Open image in new tab
 Open image in new tab
Open image in new tab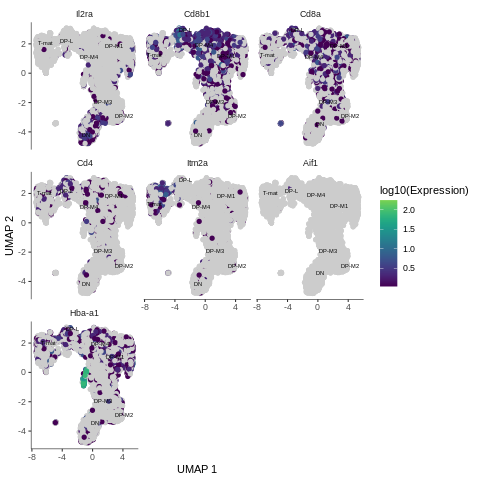 Open image in new tab
Open image in new tabOpen image in new tab
 Open image in new tab
Open image in new tab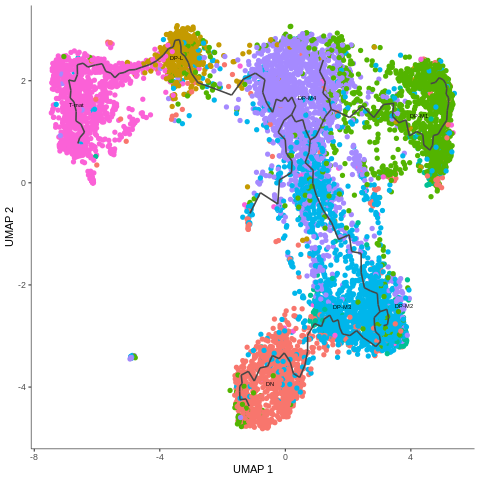 Open image in new tab
Open image in new tabOpen image in new tab
 Open image in new tab
Open image in new tab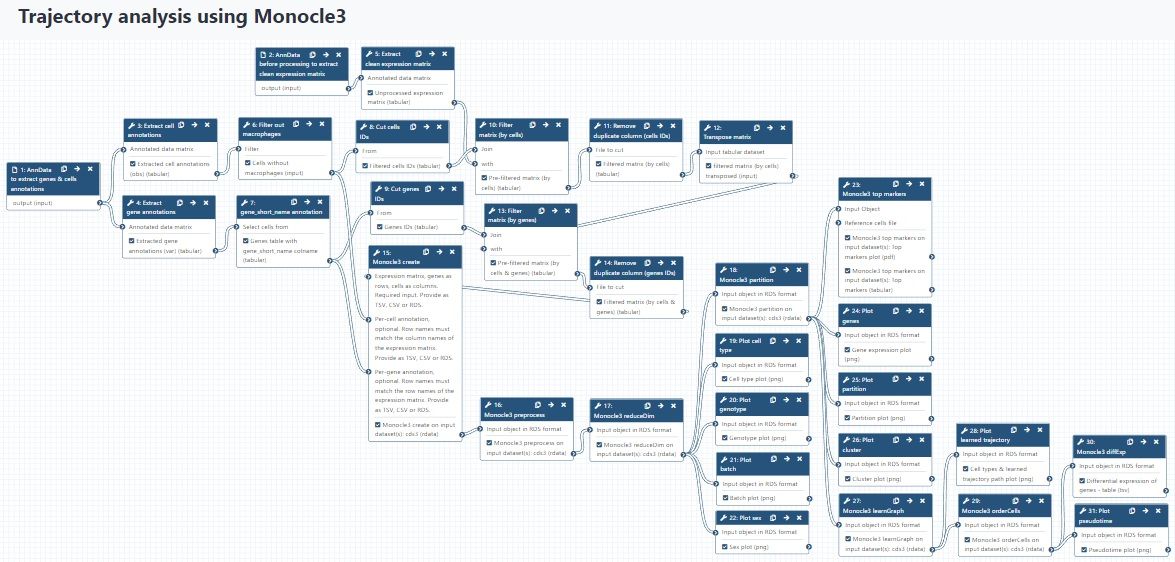 Open image in new tab
Open image in new tab
