Detection and quantitation of N-termini (degradomics) via N-TAILS
| Author(s) |
|
| Reviewers |
|
OverviewQuestions:
Objectives:
How can protein N-termini be enriched for LC-MS/MS?
How to analyze the LC-MS/MS data?
Requirements:
Run an N-TAILS data analysis.
Time estimation: 1 hourLevel: Intermediate IntermediateSupporting Materials:Published: Feb 14, 2017Last modification: Apr 8, 2025License: Tutorial Content is licensed under Creative Commons Attribution 4.0 International License. The GTN Framework is licensed under MITpurl PURL: https://gxy.io/GTN:T00226rating Rating: 1.0 (0 recent ratings, 1 all time)version Revision: 16
N-Tails is a special Proteomics technique to analyze peptide abundancy changes of protein N-termini. Prior to the MS measurement, N-Tails enriches unmodified, as well as acetylated N-termini. Both common and “unusual” N-termini are identified, where “unusual” means that the protein N-terminus was changes. This is best explained by an example: directly after translation, a protein has exactly one N-terminus. When a protease is cutting the protein in half, each half has its own N-terminus. While the N-terminus of the first half protein is exactly the same as the one of the full protein precursor (“native N-terminus”), the N-terminus of the second half is different (“neo-N-terminus”) and depends on the amino acid sequence where the protein was cut. The N-Tails technique includes the use of heavy isotope dimethyl labelling.
The figure below illustrates the mechanism of N-Tails. It was originally published by Stefan Tholen (doctoral thesis, not available online). Further reading on N-Tails and other N-terminal techniques, see Tholen et al. 2013.
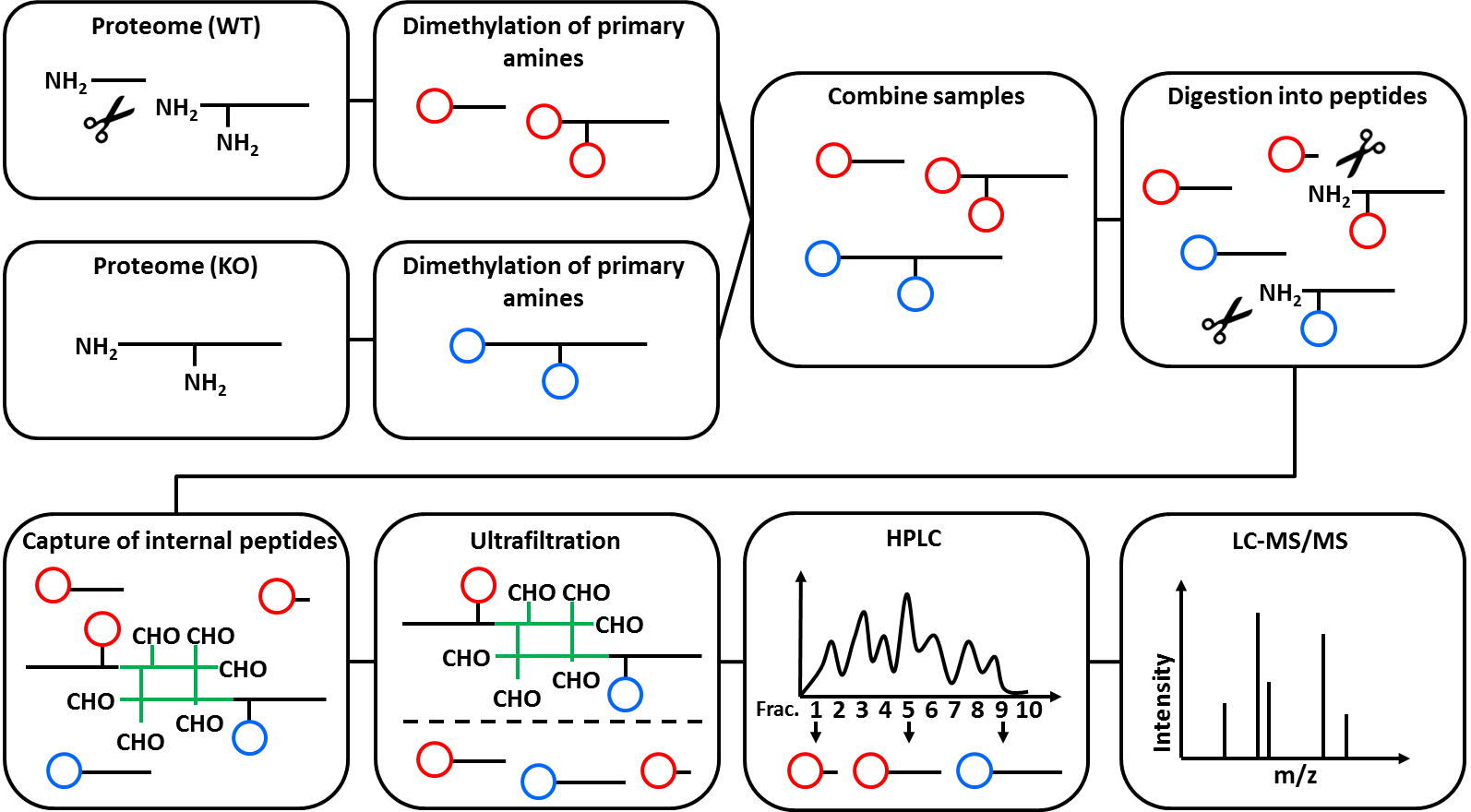 Open image in new tab
Open image in new tabThe N-Tails technique was originally designed to research protease biology and has most often been used in this field. It was originally published in Kleifeld et al. 2010.
Comment: Interpretation of N-Tails resultsBe careful when interpreting the results of N-Tails experiments. While the technique is fit to identify direct protease substrates, it does not discriminate direct from indirect (“downstream”) effects. Thus, most of the identified N-termini will not be direct protease substrates, even if their change in protein abundance is statistically significant. To identify direct protease substrates, you have to further validate substrate candidates by comparing the prime and non-prime amino acids of each identified N-terminus with the protease cleavage motif. The information can be extracted from the peptide IDs, but this step is so far not included in the workflow.
Lacking discrimination between direct and indirect effects is a general restriction also in other N-terminal screening techniques (e.g. COFRADIC), and is not specific for the N-Tails technique.
This workflow was originally built in the OpenMS framework “TOPPAS” and published in Lai, Weisser et al., MCP, 2016. It was converted to OpenMS v2.1, rebuild for the Galaxy framework and tested on the original dataset by Melanie Föll. It was designed for data analysis of a three samples combined in one MS run, a technique based on dimethyl stable isotope labeling (SIL). For more information on SIL, consult this tutorial. The original data were generated using pre-fractionation. Thus, peptides of one biological experiment are measured in multiple consecutive MS runs (one run per fraction).
The figure below gives an overview of the used Galaxy nodes. For further description of the workflow, please consider the original publication.
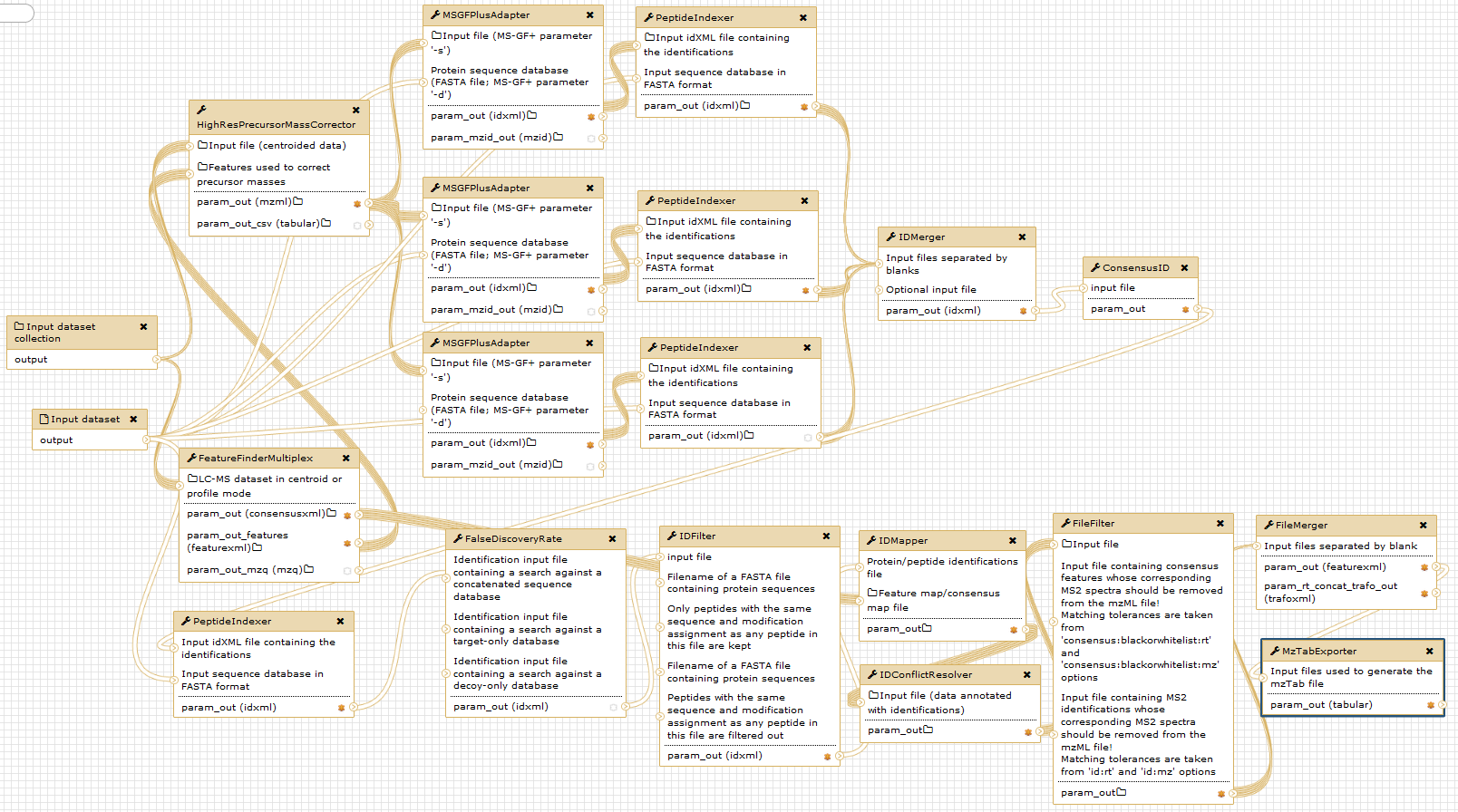 Open image in new tab
Open image in new tabNotice that the given digestion enzyme is “ArgC”, even if the proteins were digested using trypsin. Due to the used labelling method prior to digestion, lysine (“K”) residues are dimethylated. Therefore, trypsin will not cut c-terminal of lysine, but only c-terminal of arginine in a N-TAILS experiment. This resembles the ArgC specificity and generally results in longer peptides Rogers and Overall, MCP, 2013.
Input
The workflow needs two input files:
1) A collection of mzML files (multiple fractions of the same experiment). 2) A FASTA protein database for the organism of interest. For more information on protein databases, consult this tutorial
Customizing the Workflow
- Running the workflow on a non-prefractionated sample: Simply use only one file as an input.
- Running the workflow on a double dimethyl labeling (only light and heavy labeling): remove the third MSGFPlusAdapter tool and the following PeptideIndexer tool . Make sure that the mass changes are correctly given in the MSGFPlusAdapter tool .
Citation
If you use this workflow directly, or any derivative of it, in work leading to a scientific publication, please cite:
Lai, Z.W., Weisser, J., Nilse, L., Costa, F., Keller, E., Tholen, M., Kizhakkedathu, J.N., Biniossek, M., Bronsert, P., and Schilling, O. (2016). Formalin-Fixed, Paraffin-Embedded Tissues (FFPE) as a Robust Source for the Profiling of Native and Protease-Generated Protein Amino Termini. Mol. Cell. Proteomics 15, 2203–2213.
You've Finished the Tutorial
Key points
N-TAILS enriches natural protein N-termini and neo-N-termini.
neo-N-termini are typically generated by protease cleavage.
N-TAILS can be used for analysis of protease cleavage.
Frequently Asked Questions
Have questions about this tutorial? Have a look at the available FAQ pages and support channelsUseful literature
Further information, including links to documentation and original publications, regarding the tools, analysis techniques and the interpretation of results described in this tutorial can be found here.
References
- Kleifeld, O., A. Doucet, U. auf dem Keller, A. Prudova, O. Schilling et al., 2010 Isotopic labeling of terminal amines in complex samples identifies protein N-termini and protease cleavage products. Nature Biotechnology 28: 281–288. 10.1038/nbt.1611
- Tholen, S., M. M. Koczorowska, Z. W. Lai, J. Dengjel, and O. Schilling, 2013 Limited and Degradative Proteolysis in the Context of Posttranslational Regulatory Networks: Current Technical and Conceptional Advances, pp. 175–216 in Proteases: Structure and Function, Springer Vienna. 10.1007/978-3-7091-0885-7_5 ISBN: 9783709108857
Feedback
Did you use this material as an instructor? Feel free to give us feedback on how it went.
Did you use this material as a learner or student? Click the form below to leave feedback.

Citing this Tutorial
- Florian Christoph Sigloch, Björn Grüning, Detection and quantitation of N-termini (degradomics) via N-TAILS (Galaxy Training Materials). https://training.galaxyproject.org/training-material/topics/proteomics/tutorials/ntails/tutorial.html Online; accessed TODAY
- Hiltemann, Saskia, Rasche, Helena et al., 2023 Galaxy Training: A Powerful Framework for Teaching! PLOS Computational Biology 10.1371/journal.pcbi.1010752
- Batut et al., 2018 Community-Driven Data Analysis Training for Biology Cell Systems 10.1016/j.cels.2018.05.012
@misc{proteomics-ntails, author = "Florian Christoph Sigloch and Björn Grüning", title = "Detection and quantitation of N-termini (degradomics) via N-TAILS (Galaxy Training Materials)", year = "", month = "", day = "", url = "\url{https://training.galaxyproject.org/training-material/topics/proteomics/tutorials/ntails/tutorial.html}", note = "[Online; accessed TODAY]" } @article{Hiltemann_2023, doi = {10.1371/journal.pcbi.1010752}, url = {https://doi.org/10.1371%2Fjournal.pcbi.1010752}, year = 2023, month = {jan}, publisher = {Public Library of Science ({PLoS})}, volume = {19}, number = {1}, pages = {e1010752}, author = {Saskia Hiltemann and Helena Rasche and Simon Gladman and Hans-Rudolf Hotz and Delphine Larivi{\`{e}}re and Daniel Blankenberg and Pratik D. Jagtap and Thomas Wollmann and Anthony Bretaudeau and Nadia Gou{\'{e}} and Timothy J. Griffin and Coline Royaux and Yvan Le Bras and Subina Mehta and Anna Syme and Frederik Coppens and Bert Droesbeke and Nicola Soranzo and Wendi Bacon and Fotis Psomopoulos and Crist{\'{o}}bal Gallardo-Alba and John Davis and Melanie Christine Föll and Matthias Fahrner and Maria A. Doyle and Beatriz Serrano-Solano and Anne Claire Fouilloux and Peter van Heusden and Wolfgang Maier and Dave Clements and Florian Heyl and Björn Grüning and B{\'{e}}r{\'{e}}nice Batut and}, editor = {Francis Ouellette}, title = {Galaxy Training: A powerful framework for teaching!}, journal = {PLoS Comput Biol} }
Funding
These individuals or organisations provided funding support for the development of this resource
Congratulations on successfully completing this tutorial!

You can use Ephemeris's
shed-tools installcommand to install the tools used in this tutorial.shed-tools install [-g GALAXY] [-a API_KEY] -t <(curl https://training.galaxyproject.org/training-material/api/topics/proteomics/tutorials/ntails/tutorial.json | jq .admin_install_yaml -r)Alternatively you can copy and paste the following YAML
--- install_tool_dependencies: true install_repository_dependencies: true install_resolver_dependencies: true tools: []