An Introduction to Genome Assembly
| Author(s) |
|
| Reviewers |
|
OverviewQuestions:
Objectives:
How do we perform a very basic genome assembly from short read data?
Requirements:
assemble some paired end reads using Velvet
examine the output of the assembly.
Time estimation: 30 minutesLevel: Introductory IntroductorySupporting Materials:Published: May 23, 2017Last modification: Jan 23, 2026License: Tutorial Content is licensed under Creative Commons Attribution 4.0 International License. The GTN Framework is licensed under MITpurl PURL: https://gxy.io/GTN:T00034rating Rating: 4.7 (3 recent ratings, 40 all time)version Revision: 30
Genome assembly with Velvet: Background
Velvet is one of a number of de novo assemblers that use short read sets as input (e.g. Illumina Reads). The assembly method is based on the manipulation of de Bruijn graphs, via the removal of errors and the simplification of repeated regions.
CommentFor information about Velvet, you can check its (nice) Wikipedia page.
For this tutorial, we have a set of reads from an imaginary Staphylococcus aureus bacterium with a miniature genome (197,394 bp). Our mutant strain read set was sequenced with the whole genome shotgun method, using an Illumina DNA sequencing instrument. From these reads, we would like to rebuild our imaginary Staphylococcus aureus bacterium via a de novo assembly of a short read set using the Velvet assembler.
We also have a sequence for a reference genome that we will use later in the tutorial.
AgendaIn this tutorial, we will deal with:
Get the data
Hands On: Getting the data
Create and name a new history for this tutorial.
To create a new history simply click the new-history icon at the top of the history panel:
Import from Zenodo or from the data library the files:
https://zenodo.org/record/582600/files/mutant_R1.fastq https://zenodo.org/record/582600/files/mutant_R2.fastq https://zenodo.org/record/582600/files/wildtype.fna
- Copy the link location (Right-click on the filename then “Copy Link Address”)
- Open the Galaxy Upload Manager
- Select Paste/Fetch Data
- Paste the link into the text field
- For the read files, change the data-type to fastqsanger
- Press Start
Change the name of the files to
mutant_R1,mutant_R2andwildtype.fna.As a default, Galaxy uses the link as the name of the new dataset. It also does not link the dataset to a database or a reference genome.
- Click on the galaxy-pencil pencil icon for the dataset to edit its attributes
- In the central panel, change the Name field
- Click the Save button
Inspect the content of a file.
- Click on the galaxy-eye (eye) icon next to the relevant history entry
- View the content of the file in the central panel
Question
- What are four key features of a FASTQ file?
- What is the main difference between a FASTQ and a FASTA file?
- Each sequence in a FASTQ file is represented by 4 lines: 1st line is the id, 2nd line is the sequence, 3rd line is not used, and 4th line is the quality of sequencing per nucleotide
- In a FASTQ file, not only are the sequences present, but information about the quality of sequencing is also included.

The reads have been sequenced from an imaginary Staphylococcus aureus bacterium using an Illumina DNA sequencing instrument. We obtained the 2 files we imported (mutant_R1 and mutant_R2)
QuestionWhy do we have 2 files here if we only sequenced the bacteria once?
- The bacteria has been sequenced using paired-end sequencing. The first file corresponds to forward reads and the second file to reverse reads.
Evaluate the input reads
Before doing any assembly, the first questions you should ask about your input reads include:
- What is the coverage of my genome?
- How good is my read set?
- Do I need to ask for a new sequencing run?
- Is it suitable for the analysis I need to do?
We will evaluate the input reads using the FastQC tool. This tool runs a standard series of tests on your read set and returns a relatively easy-to-interpret report. We will use it to evaluate the quality of our FASTQ files and combine the results with MultiQC.
Hands On: FastQC on a fastq file
- FastQC ( Galaxy version 0.73+galaxy0) with the following parameters:
- param-files “Raw read data from your current history”:
mutant_R1.fastqandmutant_R2.fastq
- Click on param-files Multiple datasets
- Select several files by keeping the Ctrl (or COMMAND) key pressed and clicking on the files of interest
- MultiQC ( Galaxy version 1.11+galaxy1) with the following parameters:
- “Results: Which tool was used to generate logs?”:
FastQC- Click “Insert FastQC output”
- “Type of FastQC output?”:
multiple datasets, select the raw data files from FastQC
MultiQC generates a webpage combining reports for FastQC on both datasets. It includes these graphs and tables:
-
General statistics
We need to know about the data for our analysis. In particular, we need to know the read lengths as it is important in setting the maximum k-mer size for an assembly.
Comment: Getting the length of sequences- Find the MultiQC output that is a webpage and click to view
- The first table shows General Statistics for the input read files
- At the top of this table, click on Configure Columns
- Make sure the box next to Length is ticked
- Close the window
- This table should now show a column for read lengths
Question- How long are the sequences?
- What is the average coverage of the genome, given our imaginary Staphylococcus aureus bacterium has a genome of 197,394 bp?
- The sequences are 150 bp long
- We have 2 x 12,480 sequences of 150 bp, so the average genome coverage is: 2 * 12480 * 150 / 197394, or approximately 19 X coverage.
-
Sequence Quality Histograms
Dips in quality near the beginning, middle or end of the reads may determine the trimming/cleanup methods and parameters to be used, or may indicate technical problems with the sequencing process/machine run.
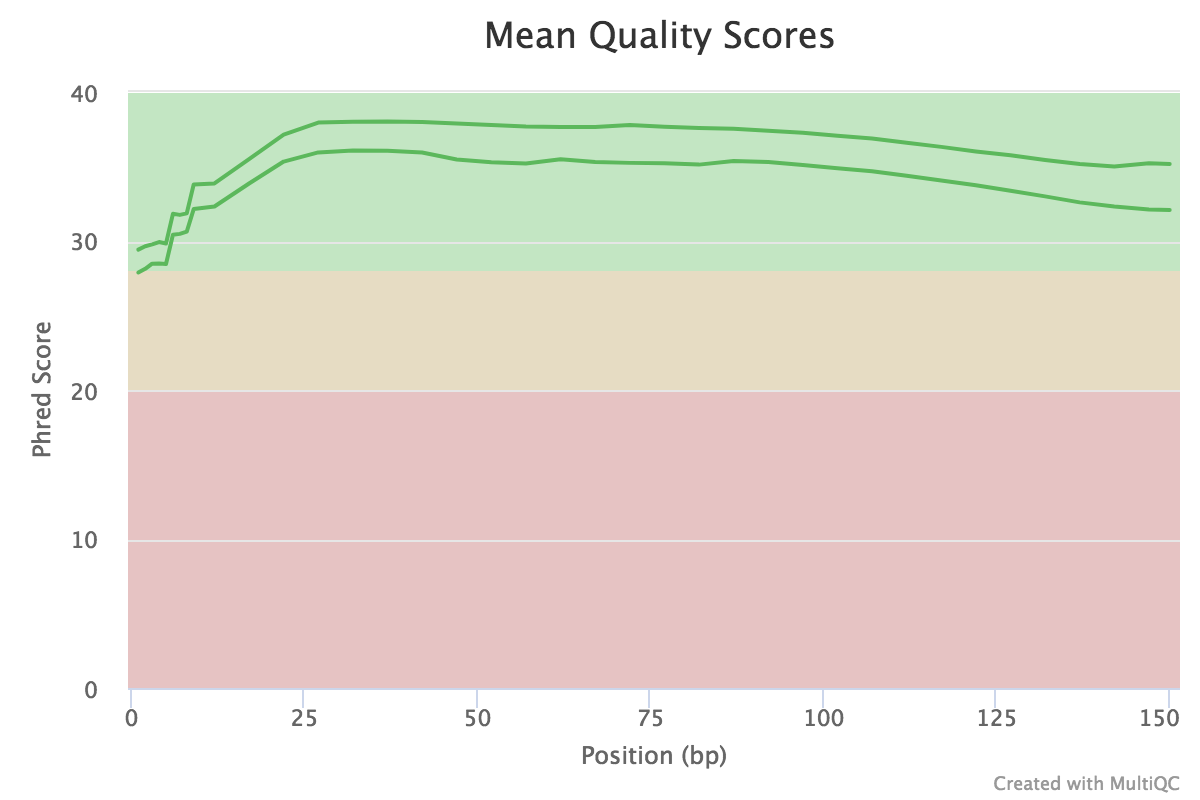 Open image in new tab
Open image in new tabFigure 1: The mean quality value across each base position in the read Question- What does the y-axis represent?
- Why is the quality score decreasing across the length of the reads?
- The y-axis represents the quality score for each base (an estimate of the error during sequencing).
- The quality score is decreasing accross the length of the reads because the sequencing become less and less reliable at the end of the reads.
-
Per Sequence GC Content
High GC organisms tend not to assemble well and may have an uneven read coverage distribution.
-
Per Base N Content
The presence of large numbers of Ns in reads may point to a poor quality sequencing run. You will need to trim these reads to remove Ns.
-
k-mer content
The presence of highly recurring k-mers may point to contamination of reads with barcodes or adapter sequences.
CommentFor a fuller discussion of FastQC outputs and warnings, see the FastQC website link, including the section on each of the output reports, and examples of “good” and “bad” Illumina data.
We won’t be doing anything to these data to clean it up as there isn’t much need. Therefore we will get on with the assembly!
Assemble reads with Velvet
Now, we want to assemble our reads to find the sequence of our imaginary Staphylococcus aureus bacterium. We will perform a de novo assembly of the reads into long contiguous sequences using the Velvet short read assembler.
The first step of the assembler is to build a de Bruijn graph. For that, it will break our reads into k-mers, i.e. fragments of length k. Velvet requires the user to input a value of k (k-mer size) for the assembly process. Small k-mers will give greater connectivity, but large k-mers will give better specificity.
Hands On: Assemble the reads
- FASTQ interlacer ( Galaxy version 1.2.0.1+galaxy0) with the following parameters:
- “Type of paired-end datasets”:
2 separate datasets- “Left-hand mates”:
mutant_R1.fastq- “Right-hand mates”:
mutant_R2.fastqCurrently our paired-end reads are in 2 files (one with the forward reads and one with the reverse reads), but Velvet requires only one file, where each read is next to its mate read. In other words, if the reads are indexed from 0, then reads 0 and 1 are paired, 2 and 3, 4 and 5, etc. Before doing the assembly per se, we need to prepare the files by combining them.
- velveth ( Galaxy version 1.2.10.3) with the following parameters:
- “Hash Length”:
29- “Input Files”
- Click on param-repeat “Input Files”
- In “1: Input Files”
- “Choose the input type”:
interleaved paired end- “read type”:
shortPaired reads- param-files “Dataset”: pairs output of FASTQ interlacer
The tool takes our reads and break them into k-mers.
- velvetg ( Galaxy version 1.2.10.2) with the following parameters:
- param-files “Velvet Dataset”: outputs of velveth
- “Using Paired Reads”:
YesThis last tool actually does the assembly.
Five files are generated. We will look at the contigs file and the stats file:
-
The Contigs file
This file contains the sequences of the contigs. In the header of each contig, a bit of information is added:
- the k-mer length (called “length”): For the value of k chosen in the assembly, a measure of how many k-mers overlap (by 1 bp each overlap) to give this length
- the k-mer coverage (called “coverage”): For the value of k chosen in the assembly, a measure of how many k-mers overlap each base position (in the assembly).
- Note that your results may look different to the example in the image below.

-
The Stats file
This is a tabular file giving for each contig the k-mer lengths, k-mer coverages and other measures. Note that your results may look different to the example in the image below.
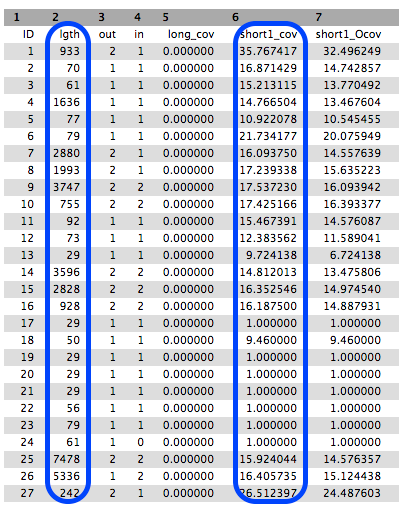
Collect some statistics on the contigs
Question
- How many contigs have been built?
- What is the mean, min and max length of the contigs?
- 306
- To compute this information, we can use the Datamash tool on the 2nd columns (length). Be careful with the first line, the header. As a result, we obtain approximately: 516 as mean, 1 as min and 8836 as max. It would mean that the smallest contig has a length of 1 bp, even smaller than k. The length on the 2nd column corresponds to length of the contig in k-mers. This means that the smallest contig has a length of 1k = 29. So to obtain the real length, we need to add k-1 to the length. We then obtain a mean contig length of 544 bp, a min contig of 29 bp and a max contig of 8864 bp.
This table is limitted, but we will now collect more basic statistics on our assembly.
Hands On: Collect fasta statistics on our contigs
- Quast ( Galaxy version 5.2.0+galaxy1) with the following parameters:
- “Assembly mode”:
Individual assembly (1 contig file per sample)- “Use customized names?”:
No- “Contigs/scaffolds file”: contigs output of velvetg
- “Type of assembly”:
Genome- “Use a reference genome?”:
Yes- “Reference genome”:
wildtype.fna- “Type of organism”:
Prokaryotes- “Lower Threshold”:
500- “Advanced options: Comma-separated list of contig length thresholds”:
0,1000
Question
- What is represented in the Icarus viewer?
- Icarus is a novel genome visualizer for accurate assessment and analysis of genomic draft assemblies. It draws contigs ordered from longest to shortest, highlights N50, N75 (NG50, NG75) and long contigs larger than a user-specified threshold
The HTML report reports many statistics computed by QUAST to assess the quality of the assembly:
- Statistics about the quality of the assembly when compared to the reference (fraction of the genome, duplication ratio, etc)
-
Misassembly statistics, including the number of misassemblies
A misassembly is a position in the contigs (breakpoints) that satisfy one of the following criteria:
- the left flanking sequence aligns over 1 kbp away from the right flanking sequence on the reference;
- flanking sequences overlap on more than 1 kbp
- flanking sequences align to different strands or different chromosomes
- Unaligned regions in the assembly
- Mismatches compared to the reference genomes
- Statistics about the assembly per se, such as the number of contigs and the length of the largest contig
Question
- How many contigs have been constructed?
- Which proportion of the reference genome do they represent?
- How many misassemblies have been found?
- Has the assembly introduced mismatches and indels?
- What are N50 and L50?
- Is there a bias in GC percentage induced by the assembly?
- 190 contigs have been constructed, but only 47 have a length > 500 bp.
- The contigs represents 87.965% of the reference genome.
- 1 misassembly has been found: it corresponds to a relocation, i.e. a misassembly event (breakpoint) where the left flanking sequence aligns over 1 kbp away from the right flanking sequence on the reference genome.
- 8.06 mismatches per 100 kbp and 4.03 indels per 100 kbp are found.
- N50 is the length for which the collection of all contigs of that length or longer covers at least half an assembly. In other words, if contigs were ordered from small to large, half of all the nucleotides will be in contigs this size or larger. And L50 is the number of contigs equal to or longer than N50: L50 is the minimal number of contigs that cover half the assembly.
- The GC % in the assembly is 33.64%, really similar to the one of the reference genome (33.43%).
Discussion
Hands On: (Optional) Rerun for values k ranging from 31 to 101
- velveth ( Galaxy version 1.2.10.3) with the same parameters as before except
- “Hash Length”: a value between 31 and 101
- velvetg ( Galaxy version 1.2.10.2) with the same parameters as before
- Quast ( Galaxy version 5.2.0+galaxy1) with the same parameters as before
We have completed an assembly on this data set for a number of k values ranging from 29 to 101. A few of the assembly metrics appear below.
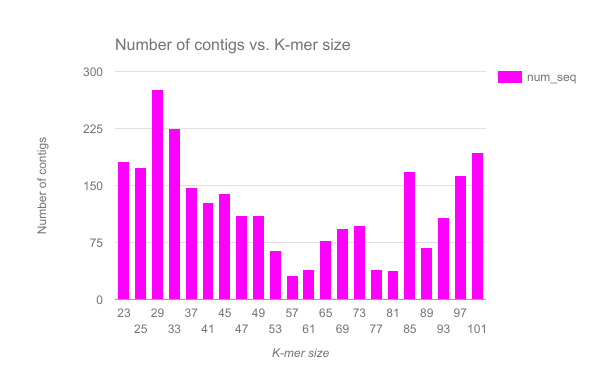 Open image in new tab
Open image in new tab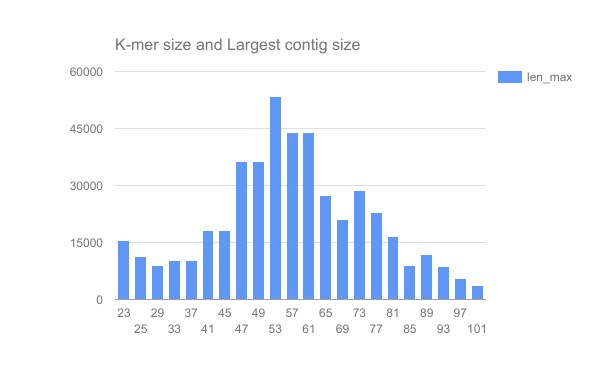 Open image in new tab
Open image in new tab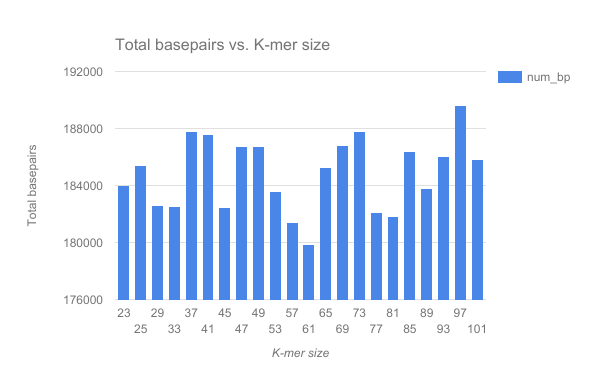 Open image in new tab
Open image in new tab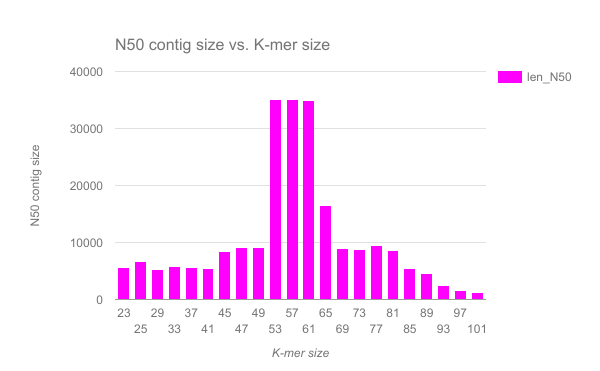 Open image in new tab
Open image in new tabQuestion
- Are there any distinct features in the charts?
- Does it look like one assembly might be better than some of the others?
The reasons for these patterns will be discussed in detail in the De Bruijn graph assembly slides and tutorial.
You've Finished the Tutorial
Key points
We assembled some Illumina fastq reads into contigs using a short read assembler called Velvet
We showed what effect one of the key assembly parameters, the k-mer size, has on the assembly
It looks as though there are some exploitable patterns in the metric data vs the k-mer size.
Frequently Asked Questions
Have questions about this tutorial? Have a look at the available FAQ pages and support channelsFeedback
Did you use this material as an instructor? Feel free to give us feedback on how it went.
Did you use this material as a learner or student? Click the form below to leave feedback.

Citing this Tutorial
- Simon Gladman, An Introduction to Genome Assembly (Galaxy Training Materials). https://training.galaxyproject.org/training-material/topics/assembly/tutorials/general-introduction/tutorial.html Online; accessed TODAY
- Hiltemann, Saskia, Rasche, Helena et al., 2023 Galaxy Training: A Powerful Framework for Teaching! PLOS Computational Biology 10.1371/journal.pcbi.1010752
- Batut et al., 2018 Community-Driven Data Analysis Training for Biology Cell Systems 10.1016/j.cels.2018.05.012
@misc{assembly-general-introduction, author = "Simon Gladman", title = "An Introduction to Genome Assembly (Galaxy Training Materials)", year = "", month = "", day = "", url = "\url{https://training.galaxyproject.org/training-material/topics/assembly/tutorials/general-introduction/tutorial.html}", note = "[Online; accessed TODAY]" } @article{Hiltemann_2023, doi = {10.1371/journal.pcbi.1010752}, url = {https://doi.org/10.1371%2Fjournal.pcbi.1010752}, year = 2023, month = {jan}, publisher = {Public Library of Science ({PLoS})}, volume = {19}, number = {1}, pages = {e1010752}, author = {Saskia Hiltemann and Helena Rasche and Simon Gladman and Hans-Rudolf Hotz and Delphine Larivi{\`{e}}re and Daniel Blankenberg and Pratik D. Jagtap and Thomas Wollmann and Anthony Bretaudeau and Nadia Gou{\'{e}} and Timothy J. Griffin and Coline Royaux and Yvan Le Bras and Subina Mehta and Anna Syme and Frederik Coppens and Bert Droesbeke and Nicola Soranzo and Wendi Bacon and Fotis Psomopoulos and Crist{\'{o}}bal Gallardo-Alba and John Davis and Melanie Christine Föll and Matthias Fahrner and Maria A. Doyle and Beatriz Serrano-Solano and Anne Claire Fouilloux and Peter van Heusden and Wolfgang Maier and Dave Clements and Florian Heyl and Björn Grüning and B{\'{e}}r{\'{e}}nice Batut and}, editor = {Francis Ouellette}, title = {Galaxy Training: A powerful framework for teaching!}, journal = {PLoS Comput Biol} }
Funding
These individuals or organisations provided funding support for the development of this resource
Congratulations on successfully completing this tutorial!
You can use Ephemeris's
shed-tools installcommand to install the tools used in this tutorial.shed-tools install [-g GALAXY] [-a API_KEY] -t <(curl https://training.galaxyproject.org/training-material/api/topics/assembly/tutorials/general-introduction/tutorial.json | jq .admin_install_yaml -r)Alternatively you can copy and paste the following YAML
--- install_tool_dependencies: true install_repository_dependencies: true install_resolver_dependencies: true tools: - name: fastq_paired_end_interlacer owner: devteam revisions: 2ccb8dcabddc tool_panel_section_label: FASTA/FASTQ tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: fastqc owner: devteam revisions: e7b2202befea tool_panel_section_label: FASTA/FASTQ tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: fastqc owner: devteam revisions: 3d0c7bdf12f5 tool_panel_section_label: FASTA/FASTQ tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: velvet owner: devteam revisions: 5da9a0e2fb2d tool_panel_section_label: Assembly tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: velvet owner: devteam revisions: 920677cd220f tool_panel_section_label: Assembly tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: velvet owner: devteam revisions: 5da9a0e2fb2d tool_panel_section_label: Assembly tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: velvet owner: devteam revisions: 920677cd220f tool_panel_section_label: Assembly tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: multiqc owner: iuc revisions: abfd8a6544d7 tool_panel_section_label: Quality Control tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: multiqc owner: iuc revisions: b2f1f75d49c4 tool_panel_section_label: Quality Control tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: quast owner: iuc revisions: 59db8ea8c845 tool_panel_section_label: Assembly tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: quast owner: iuc revisions: 72472698a2df tool_panel_section_label: Assembly tool_shed_url: https://toolshed.g2.bx.psu.edu/