name: inverse layout: true class: center, middle, inverse <div class="my-header"><span> <a href="/training-material/topics/assembly" title="Return to topic page" ><i class="fa fa-level-up" aria-hidden="true"></i></a> <a href="https://github.com/galaxyproject/training-material/edit/main/topics/assembly/tutorials/general-introduction/slides.html"><i class="fa fa-pencil" aria-hidden="true"></i></a> </span></div> <div class="my-footer"><span> <img src="/training-material/assets/images/GTN-60px.png" alt="Galaxy Training Network" style="height: 40px;"/> </span></div> --- <img src="/training-material/assets/images/GTNLogo1000.png" alt="Galaxy Training Network" class="cover-logo"/> <br/> <br/> # An Introduction to Genome Assembly <br/> <br/> <div markdown="0"> <div class="contributors-line"> <ul class="text-list"> <li> <a href="/training-material/hall-of-fame/slugger70/" class="contributor-badge contributor-slugger70"><img src="/training-material/assets/images/orcid.png" alt="orcid logo" width="36" height="36"/><img src="https://avatars.githubusercontent.com/slugger70?s=36" alt="Simon Gladman avatar" width="36" class="avatar" /> Simon Gladman</a></li> </ul> </div> </div> <!-- modified date --> <div class="footnote" style="bottom: 8em;"> <i class="far fa-calendar" aria-hidden="true"></i><span class="visually-hidden">last_modification</span> Updated: <i class="fas fa-fingerprint" aria-hidden="true"></i><span class="visually-hidden">purl</span><abbr title="Persistent URL">PURL</abbr>: <a href="https://gxy.io/GTN:S00033">gxy.io/GTN:S00033</a> </div> <!-- other slide formats (video and plain-text) --> <div class="footnote" style="bottom: 5em;"> <i class="fas fa-file-alt" aria-hidden="true"></i><span class="visually-hidden">text-document</span><a href="slides-plain.html"> Plain-text slides</a> | <div class="btn-group"> <!-- dropdown with all recordings --> <a href="/training-material/topics/assembly/tutorials/general-introduction/recordings/" class="btn btn-default dropdown-toggle topic-icon" data-toggle="dropdown" aria-expanded="false" title="Latest recordings of this material in the GTN Video Library"> <i class="fas fa-video" aria-hidden="true"></i><span class="visually-hidden">video</span> Recordings </a> <ul class="dropdown-menu"> <li><a class="dropdown-item" href="/training-material/topics/assembly/tutorials/general-introduction/recordings/index.html#lecture-recording-15-february-2021" title="View the recording for this tutorial"> <i class="fas fa-video" aria-hidden="true"></i><span class="visually-hidden">video</span> Lecture (February 2021) - 25m</a> </li> <li><a class="dropdown-item" href="/training-material/topics/assembly/tutorials/general-introduction/recordings/" title="View all recordings for this tutorial"> <i class="fas fa-video" aria-hidden="true"></i><span class="visually-hidden">video</span> View All</a> </li> </ul> </div> </div> <!-- usage tips --> <div class="footnote" style="bottom: 2em;"> <strong>Tip: </strong>press <kbd>P</kbd> to view the presenter notes | <i class="fa fa-arrows" aria-hidden="true"></i><span class="visually-hidden">arrow-keys</span> Use arrow keys to move between slides </div> ??? Presenter notes contain extra information which might be useful if you intend to use these slides for teaching. Press `P` again to switch presenter notes off Press `C` to create a new window where the same presentation will be displayed. This window is linked to the main window. Changing slides on one will cause the slide to change on the other. Useful when presenting. --- ## Requirements Before diving into this slide deck, we recommend you to have a look at: - [Introduction to Galaxy Analyses](/training-material/topics/introduction) - [Sequence analysis](/training-material/topics/sequence-analysis) - Quality Control: [<i class="fab fa-slideshare" aria-hidden="true"></i><span class="visually-hidden">slides</span> slides](/training-material/topics/sequence-analysis/tutorials/quality-control/slides.html) - [<i class="fas fa-laptop" aria-hidden="true"></i><span class="visually-hidden">tutorial</span> hands-on](/training-material/topics/sequence-analysis/tutorials/quality-control/tutorial.html) --- ### <i class="far fa-question-circle" aria-hidden="true"></i><span class="visually-hidden">question</span> Questions - How do we perform a very basic genome assembly from short read data? --- ### <i class="fas fa-bullseye" aria-hidden="true"></i><span class="visually-hidden">objectives</span> Objectives - assemble some paired end reads using Velvet - examine the output of the assembly. --- .enlarge120[ # ***De novo* Genome Assembly** ] #### With thanks to T Seemann, D Bulach, I Cooke and Simon Gladman --- .enlarge120[ # ***De novo* assembly** ] .pull-left[ **The process of reconstructing the original DNA sequence from the fragment reads alone.** * Instinctively like a jigsaw puzzle * Find reads which "fit together" (overlap) * Could be missing pieces (sequencing bias) * Some pieces will be dirty (sequencing errors) ] .pull-right[  ] --- # **Another View** 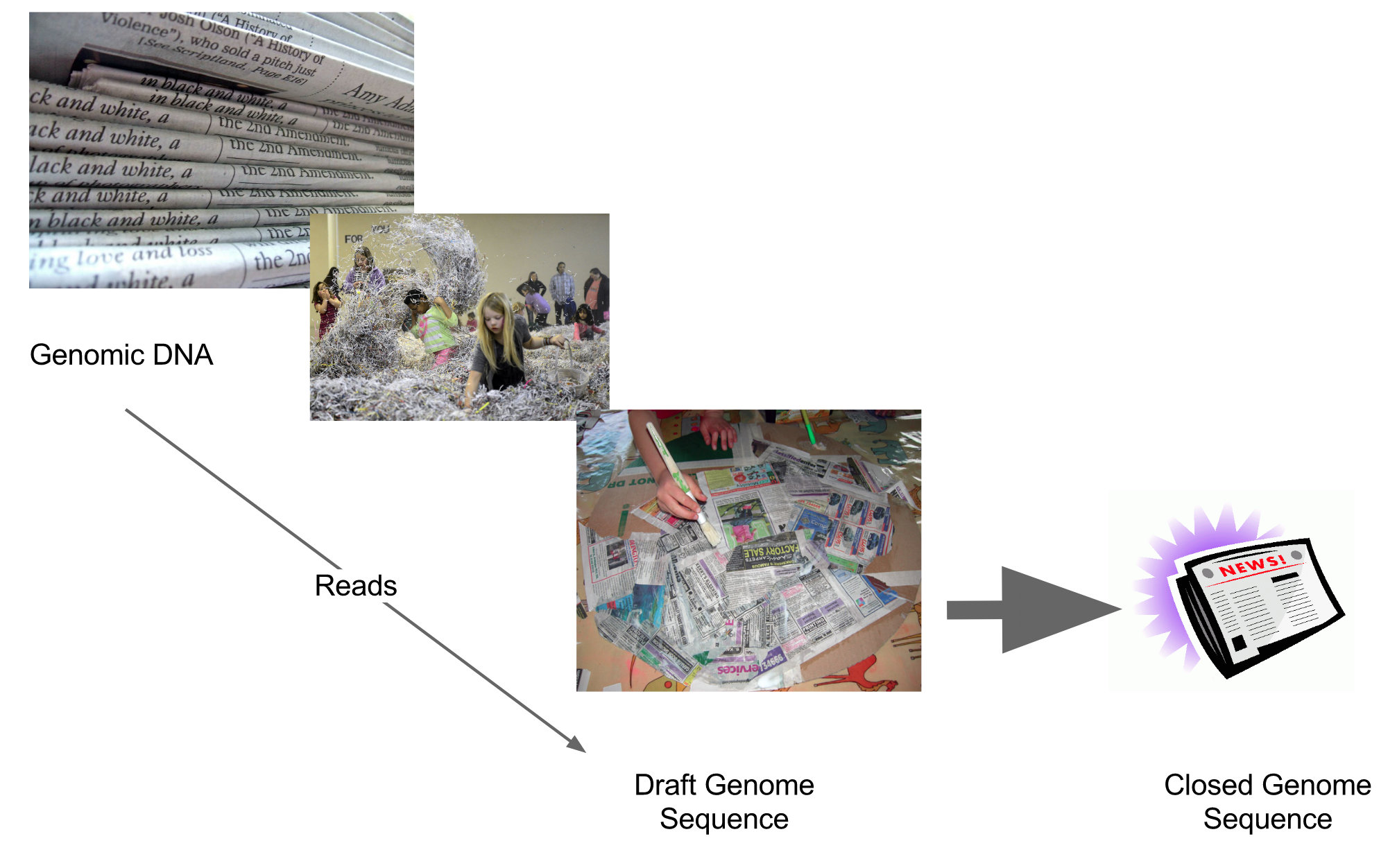 --- # **Assembly: An Example** --- # **A small "genome"**  --- # **Shakespearomics**  --- # **Shakespearomics**  --- # **Shakespearomics**  --- # **So far, so good!** --- # **The Awful Truth**  ## "Genome assembly is impossible." - A/Prof. Mihai Pop --- .enlarge120[ # **Why is it so hard?** ] .pull-left[ * Millions of pieces * Much, much shorter than the genome * Lots of them look similar * Missing pieces * Some parts can't be sequenced easily * Dirty Pieces * Lots of errors in reads ] .pull-right[  ] --- # **Assembly recipe** * Find all overlaps between reads * Hmm, sounds like a lot of work.. * Build a graph * A picture of the read connections * Simplify the graph * Sequencing errors will mess it up a lot * Traverse the graph * Trace a sensible path to produce a consensus --- 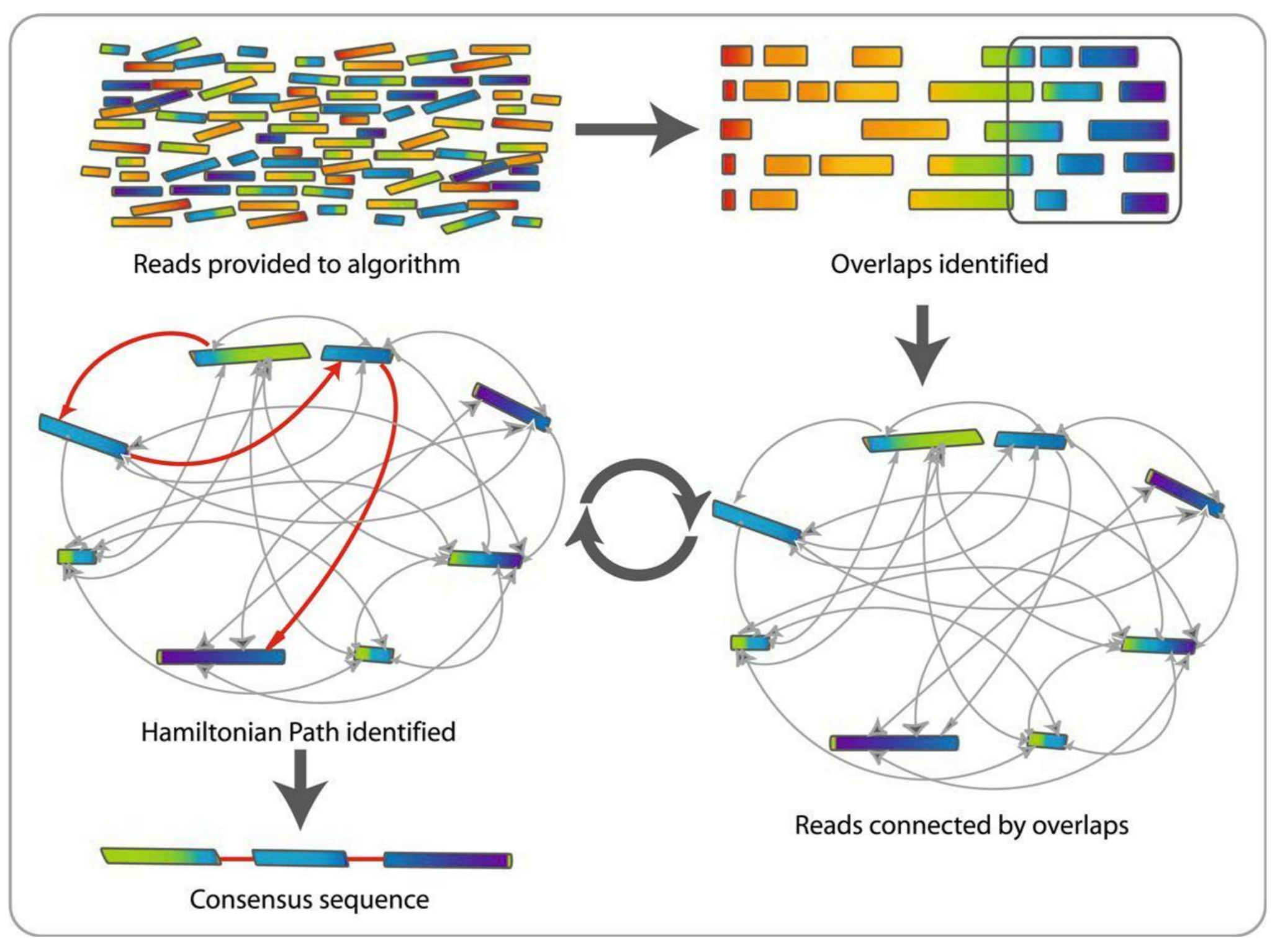 --- # **A more realistic graph** 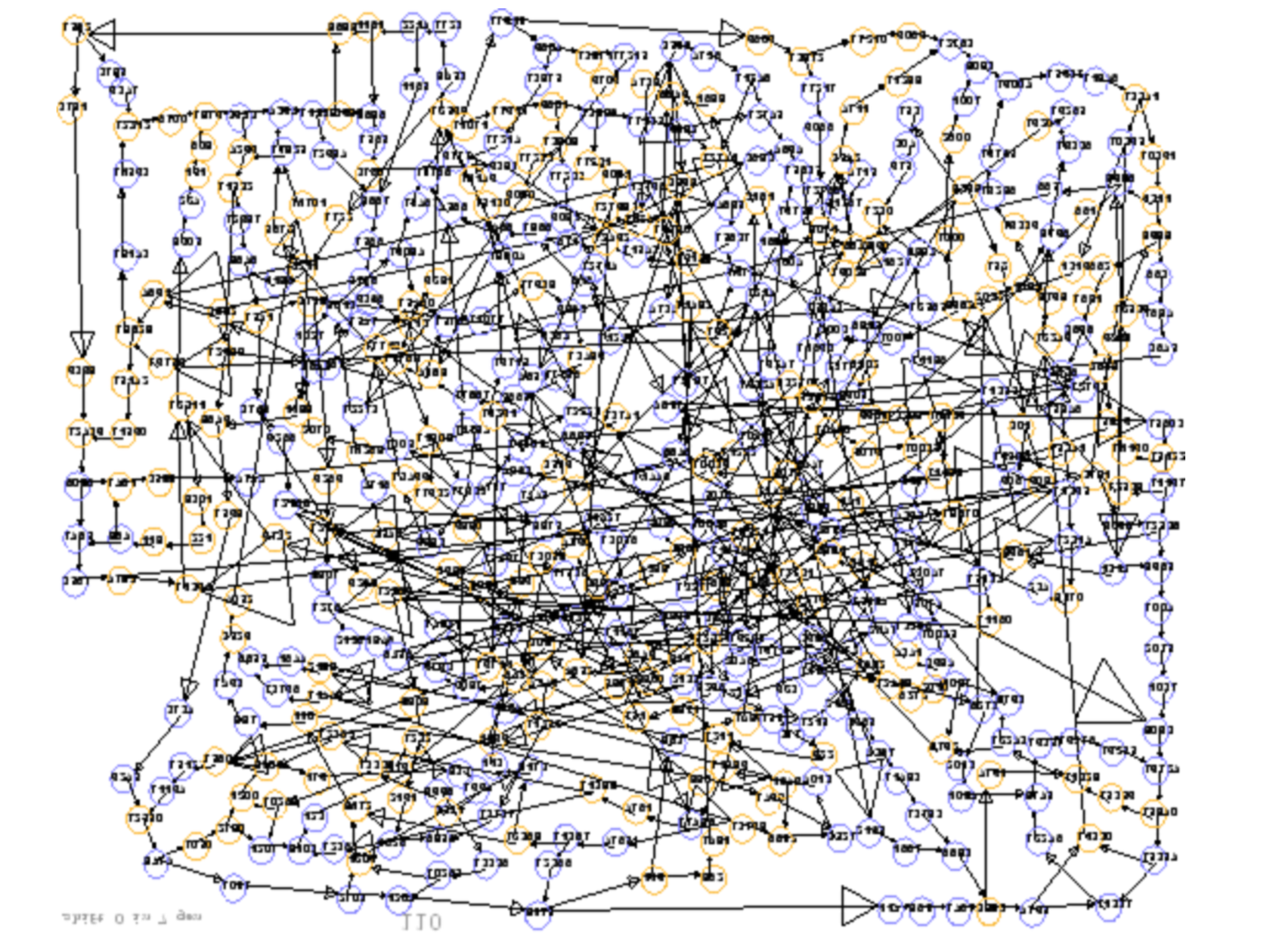 --- # .image-15[] **What ruins the graph?** * Read errors * Introduces false edges and nodes * Non haploid organisms * Heterozygosity causes lots of detours * Repeats * If they are longer than the read length * Causes nodes to be shared, locality confusion. --- # **Repeats** --- .enlarge120[ # **What is a repeat?** ] .pull-left[ #### ***A segment of DNA which occurs more than once in the genome sequence*** * Very common * Transposons (self replicating genes) * Satellites (repetitive adjacent patterns) * Gene duplications (paralogs) ] .pull-right[  ] --- # **Effect on Assembly** 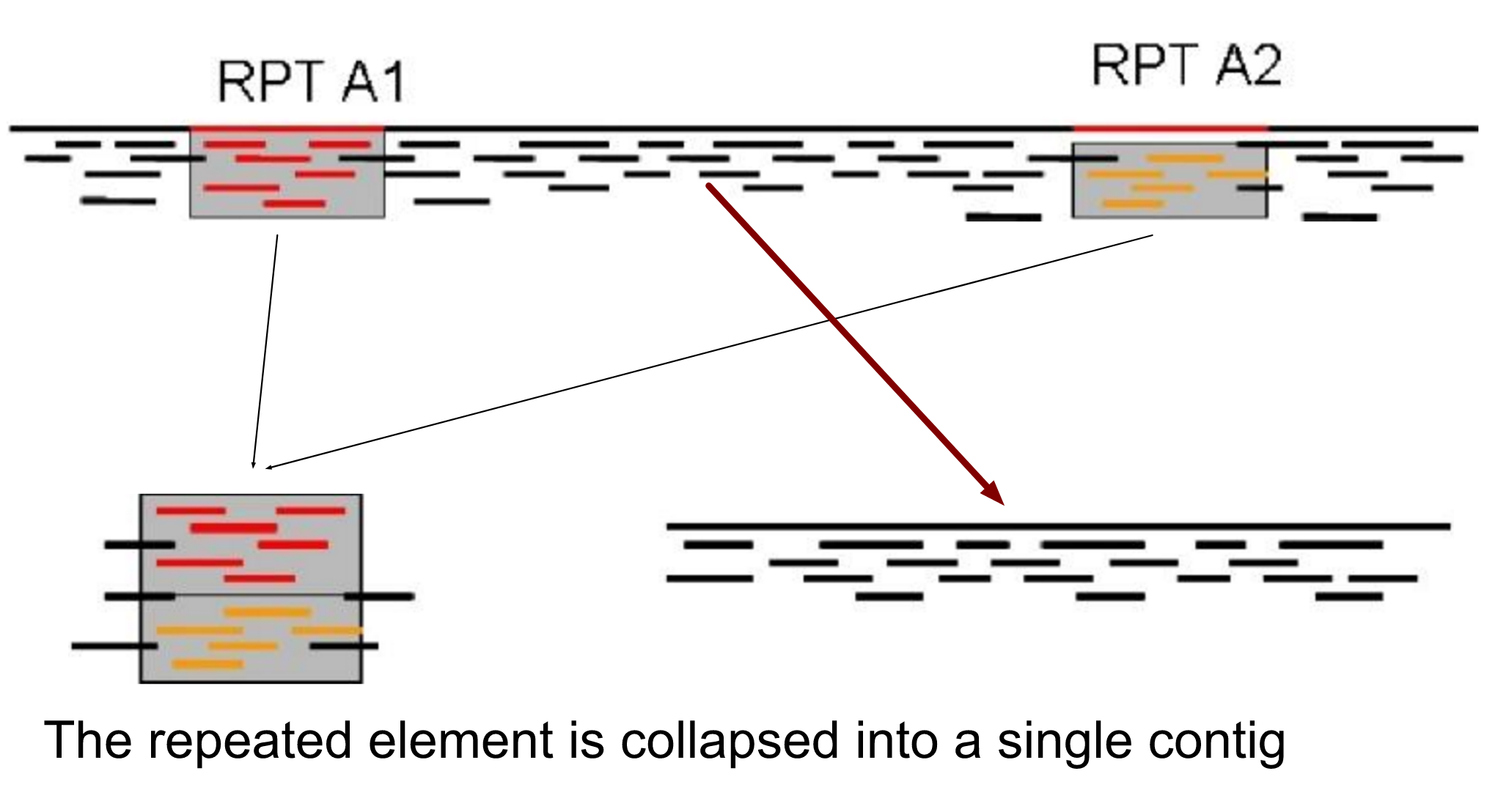 --- .enlarge120[ # **The law of repeats** .image-15[] ] ## **It is impossible to resolve repeats of length S unless you have reads longer than S** ## **It is impossible to resolve repeats of length S unless you have reads longer than S** --- # **Scaffolding** --- .enlarge120[ # **Beyond contigs** ] .pull-left[ Contig sizes are limited by: * the length of the repeats in your genome * Can't change this * the length (or "span") of the reads * Use long read technology * Use tricks with other technology ] --- .enlarge120[ # **Types of reads** ] .pull-left[.enlarge120[**Example fragment**]] .remark-code[.enlarge120[atcgtatgatcttgagattctctcttcccttatagctgctata]] .pull-left[.enlarge120[**"Single-end" read**]] .remark-code[.enlarge120[**atcgtatg**atcttgagattctctcttcccttatagctgctata]] sequence *one* end of the fragment .pull-left[.enlarge120[**"Paired-end" read**]] .remark-code[.enlarge120[**atcgtatg**atcttgagattctctcttcccttatag**ctgctata**]] sequence both ends of the same fragment **We can exploit this information!** --- .enlarge120[# **Scaffolding**] * **Paired end reads** * Known sequences at each end of fragment * Roughly known fragment length * **Most ends will occur in same contig** * **Some will occur in different contigs** * ***evidence that these contigs are linked*** --- .enlarge120[# **Contigs to Scaffolds**] 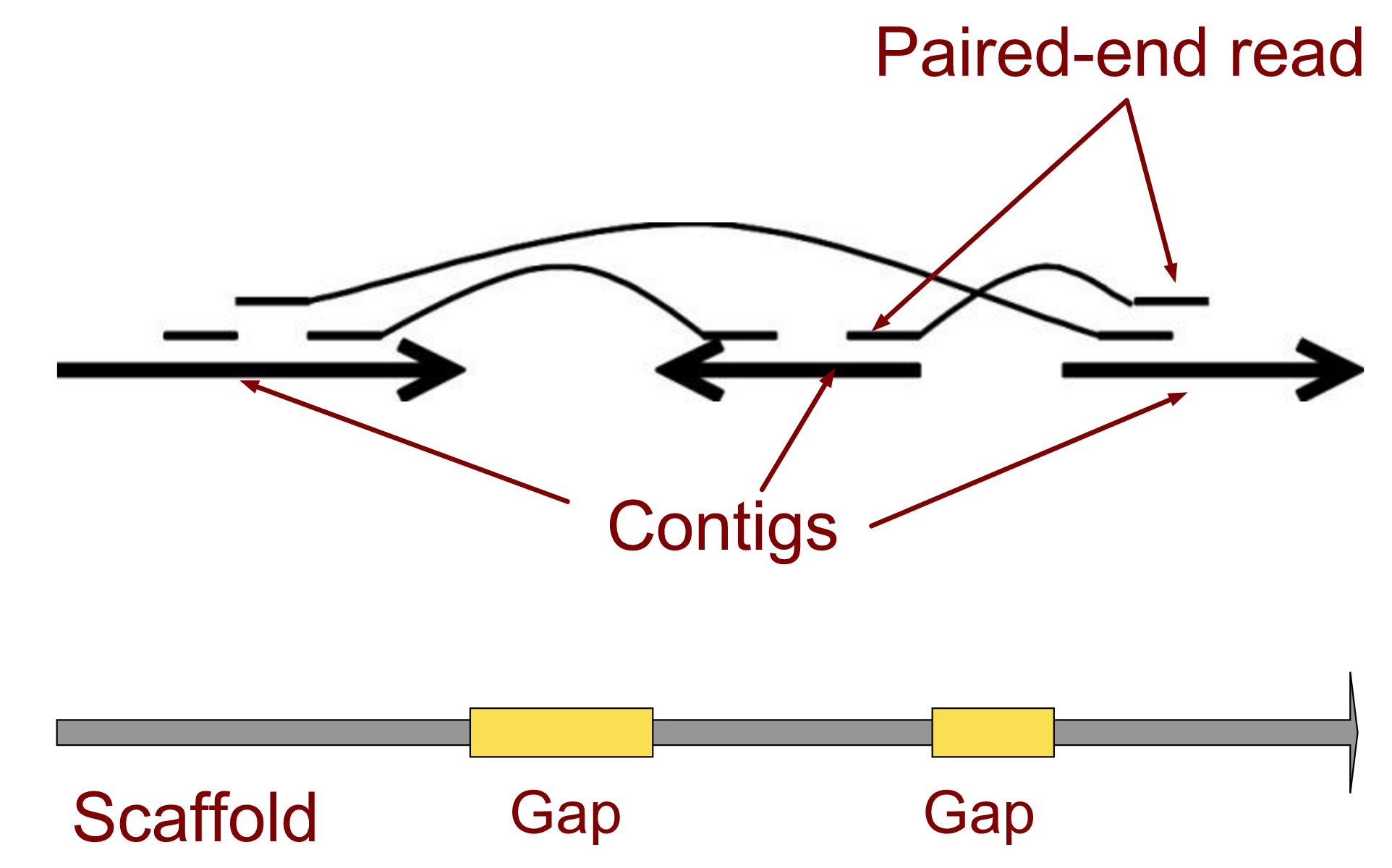 --- .enlarge120[# **Assessing assemblies**] * We desire * Total length similar to genome size * Fewer, larger contigs * Correct contigs * Metrics * No generally useful measure. (No real prior information) * Longest contigs, total base pairs in contigs, **N50**, ... --- .enlarge120[# **The "N50"**] .enlarge120[***The length of that contig from which 50% of the bases are in it and shorter contigs***] * Imagine we have 7 contigs with lengths: * 1, 1, 3, 5, 8, 12, 20 * Total * 1+1+3+5+8+12+20 = 50 * N50 is the "halfway sum" = 25 * 1+1+3+5+8+**12** = 30 (>25) so **N50 is 12** --- .enlarge120[# **2 levels of assembly**] * Draft assembly * Will contain a number of non-linked scaffolds with gaps of unknown sequence * Fairly easy to get to * Closed (finished) assembly * One sequence for each chromosome * Takes a **lot** more work * Small genomes are becoming easier with long read tech * Large genomes are the province of big consortia (e.g. Human Genome Consortium) --- .enlarge120[# **How do I do it?**] --- .enlarge120[ # **Example** * Culture your bacterium * Extract your genomic DNA * Send it to your sequencing centre for Illumina sequencing * 250bp paired end * Get back 2 files * .remark-code[MRSA_R1.fastq.gz] * .remark-code[MRSA_R2.fastq.gz] * ***Now what?*** ] --- .enlarge120[# **Assembly tools** * **Genome** * **Velvet, Velvet Optimizer, Spades,** Abyss, MIRA, Newbler, SGA, AllPaths, Ray, SOAPdenovo, ... * Meta-genome * Meta Velvet, SGA, custom scripts + above * Transcriptome * Trinity, Oases, Trans-abyss ***And many, many others...*** ] --- .enlarge120[ # **Assembly Exercise #1** * We will do a simple assembly using **Velvet** in **Galaxy** * We can do a number of different assemblies and compare some assembly metrics. ] --- ### <i class="fas fa-key" aria-hidden="true"></i><span class="visually-hidden">keypoints</span> Key points - We assembled some Illumina fastq reads into contigs using a short read assembler called Velvet - We showed what effect one of the key assembly parameters, the k-mer size, has on the assembly - It looks as though there are some exploitable patterns in the metric data vs the k-mer size. --- ## Thank You! This material is the result of a collaborative work. Thanks to the [Galaxy Training Network](https://training.galaxyproject.org) and all the contributors! <div markdown="0"> <div class="contributors-line"> <table class="contributions"> <tr> <td><abbr title="These people wrote the bulk of the tutorial, they may have done the analysis, built the workflow, and wrote the text themselves.">Author(s)</abbr></td> <td> <a href="/training-material/hall-of-fame/slugger70/" class="contributor-badge contributor-slugger70"><img src="/training-material/assets/images/orcid.png" alt="orcid logo" width="36" height="36"/><img src="https://avatars.githubusercontent.com/slugger70?s=36" alt="Simon Gladman avatar" width="36" class="avatar" /> Simon Gladman</a> </td> </tr> <tr class="reviewers"> <td><abbr title="These people reviewed this material for accuracy and correctness">Reviewers</abbr></td> <td> <a href="/training-material/hall-of-fame/hexylena/" class="contributor-badge contributor-badge-small contributor-hexylena"><img src="https://avatars.githubusercontent.com/hexylena?s=36" alt="Helena Rasche avatar" width="36" class="avatar" /></a><a href="/training-material/hall-of-fame/nsoranzo/" class="contributor-badge contributor-badge-small contributor-nsoranzo"><img src="https://avatars.githubusercontent.com/nsoranzo?s=36" alt="Nicola Soranzo avatar" width="36" class="avatar" /></a><a href="/training-material/hall-of-fame/shiltemann/" class="contributor-badge contributor-badge-small contributor-shiltemann"><img src="https://avatars.githubusercontent.com/shiltemann?s=36" alt="Saskia Hiltemann avatar" width="36" class="avatar" /></a><a href="/training-material/hall-of-fame/gallardoalba/" class="contributor-badge contributor-badge-small contributor-gallardoalba"><img src="https://avatars.githubusercontent.com/gallardoalba?s=36" alt="Cristóbal Gallardo avatar" width="36" class="avatar" /></a><a href="/training-material/hall-of-fame/willdurand/" class="contributor-badge contributor-badge-small contributor-willdurand"><img src="https://avatars.githubusercontent.com/willdurand?s=36" alt="William Durand avatar" width="36" class="avatar" /></a><a href="/training-material/hall-of-fame/njall/" class="contributor-badge contributor-badge-small contributor-njall"><img src="https://avatars.githubusercontent.com/njall?s=36" alt="Niall Beard avatar" width="36" class="avatar" /></a><a href="/training-material/hall-of-fame/bebatut/" class="contributor-badge contributor-badge-small contributor-bebatut"><img src="https://avatars.githubusercontent.com/bebatut?s=36" alt="Bérénice Batut avatar" width="36" class="avatar" /></a></td> </tr> </table> </div> </div> <div style="display: flex;flex-direction: row;align-items: center;justify-content: center;"> <img src="/training-material/assets/images/GTNLogo1000.png" alt="Galaxy Training Network" style="height: 100px;"/> </div> Tutorial Content is licensed under <a rel="license" href="http://creativecommons.org/licenses/by/4.0/">Creative Commons Attribution 4.0 International License</a>.<br/>