Generic plugins
| Author(s) |
|
| Reviewers |
|
OverviewQuestions:
Objectives:
How can visualization plugins benefit science?
Requirements:
Implement a first Galaxy visualization
Understand the client side vs. server side principle
- Javascript knowledge
Time estimation: 1 hour 30 minutesSupporting Materials:Published: Sep 30, 2016Last modification: Nov 9, 2023License: Tutorial Content is licensed under Creative Commons Attribution 4.0 International License. The GTN Framework is licensed under MITpurl PURL: https://gxy.io/GTN:T00121version Revision: 21
Visualizations may be very helpful in understanding data better. There is a whole range of visualizations, from rather simple scatter and barplots up to projections of high dimensional data or even entire genomes. Many of these visualizations often require a lot of tweaking and changes in settings like zooming in and assigning colors, etc. Therefore, visualizations are ideally interactive, and changing settings is often an initial step in exploring data. For this reason it may be inconvenient to make use of static galaxy tools because it lacks these interactive features. For these situations Galaxy offers the option to create visualizations plugins, file format specific javascripts that integrate with the history menu, without making redundant copies of data.
In this tutorial we shall go through how this system works and create a simple visualization plugin. The tool will create a visualization of the number of aligned reads per chromosome of a BAM file, and we will discuss possible optimizations and advantages and disadvantages of the proposed implementation.
If you want to make visualizations ready for production, it is essential to have a good understanding of HTML5 and JavaScript as these are the basic languages in which they are written. However, for this tutorial we will keep it basic.
Additional documentation about Galaxy visualizations can be found here:
AgendaIn this tutorial, we will deal with:
Part 1
The visualization we are going to create in this tutorial, is a tool that shows the number of aligned
reads per chromosome of a BAM file. The first thing we need to do is to come up with a name.
Let’s call it alignment_rname_boxplot. Note that the reference sequences (usually chromosomes)
to which we align are named RNAME in the BAM/SAM specification.
The development of a Galaxy visualization takes place within the Galaxy codebase.
Hands On: Data upload
- Clone an instance of Galaxy in a path, further referred to as
$GALAXY_ROOTExplore the plugin directory as follows:
$ cd $GALAXY_ROOT/config/plugins/visualizationsCreate a new directory for our new plugin project
$ mkdir alignment_rname_boxplot $ cd alignment_rname_boxplotMake three (sub-)directories to complete the structure of the project:
$ mkdir config $ mkdir static $ mkdir templates
Linking the plugin with Galaxy
To create a bridge between our not-yet-written plugin and Galaxy, we need to write a configuration in XML format.
Hands On: Data uploadCreate the file
config/alignment_rname_boxplot.xmlwith the following contents:<?xml version="1.0" encoding="UTF-8"?> <!DOCTYPE visualization SYSTEM "../../visualization.dtd"> <visualization name="alignment_rname_boxplot"> <data_sources> <data_source> <model_class>HistoryDatasetAssociation</model_class> <test type="isinstance" test_attr="datatype" result_type="datatype">binary.Bam</test> <test type="isinstance" test_attr="datatype" result_type="datatype">tabular.Sam</test> <to_param param_attr="id">dataset_id</to_param> </data_source> </data_sources> <params> <param type="dataset" var_name_in_template="hda" required="true">dataset_id</param> </params> <template>alignment_rname_boxplot.mako</template> </visualization>
This configures the plugin’s name, which shall appear on pressing the visualization button in the history menu. It also links the plugin to two file formats: BAM and SAM, which means that for any history item of these file formats the plugin will automatically become available.
It also includes a reference to a mako template file (HTML + Python syntax), to be found in the
templates directory (we will create this file in the next section). The var_name_in_template
parameter is set to the value hda, which will be the name of the variable in the mako template
corresponding to the dataset to be visualized.
Creating the visualization
We have linked our visualization to a mako file (which we have not yet created). This file is a blueprint for the visualization. This means that for every invocation of the visualization, the mako file will be compiled to render an HTML file.
Beause we would like the visualization to load quickly, computationally intensive tasks should not be done prior to loading. A bit of server-side rendering in itself is not a problem, but the visualizations (written in HTML and/or JS) should do most of the actual calculations and conversions on the client side (in the browser). Therefore, unlike regular Galaxy tools, parsing files does not take place on the server, but instead data will be downloaded by the client via an exposed Galaxy URL prior to client-side rendering.
The most basic part of the mako file are the variables used for further web development, given below.
<!DOCTYPE HTML>
<%
import os
## Generates hash (hdadict['id']) of history item
hdadict = trans.security.encode_dict_ids( hda.to_dict() )
## Finds the parent directory of galaxy (/, /galaxy, etc.)
root = h.url_for( '/' )
## Determines the exposed URL of the ./static directory
app_root = root + 'plugins/visualizations/'+visualization_name+'/static/'
## Actual file URL:
file_url = os.path.join(root, 'datasets', hdadict['id'], "display?to_ext="+hda.ext)
%>
hdadictis a variable that contains a file identifier that has been encoded to it’s exposed uid.rootindicates location of Galaxy on the webserver (e.g./,/galaxy/,/galaxy-pub/, etc).app_rootcontains the exposed url of the static files for this visualization.file_urlcontains the exposed url of the dataset selected (by user) for visualization.
We could obtain the BAM file client-side by downloading the BAM/SAM file in its entirety via file_url. However, BAM files can become quite large and it is usually not desired to transfer such datasets over the network. In our case it is also rather inconvenient to parse the BAM file with Javascript, just to count the number of reads.
Fortunately, BAM files have indices. These indices are brief summaries describing the number of entries per chromosome, in order to be able to access the data contained in them more quickly. In the mako template we can access a BAM index as hda.metadata.bam_index. (Note that this is a file path on the server, not an exposed URL).
Samtools has a command named idxstats which is able to leverage this BAM index you. However,
since visualizations do not have dependency management, it is very tricky to let the mako template
do a system call to samtools. Fortunately, the Galaxy ecosystem ships with a built-in pysam
dependency, a library that can do any native samtools command within python.
The *.metadata.bam_index is a special kind of file in the Galaxy ecosystem. It is actually an
invisible file in Galaxy, linked to another history item, but does have a unique filename.
So, for the BAM file ./database/files/000/dataset_001.dat, our BAI file (index) is not
./database/files/000/dataset_001.dat.bai but could be ./database/files/000/dataset_002.dat or
./database/files/000/dataset_003.dat. We can create a symlink to this index file to ensure the
bam file and its index share the same prefix, as expected by samtools and pysam.
We can create this symlink as follows in our mako template:
## Ensure BAI index is symlinked
bai_target = hda.file_name+'.bai'
import os
if not os.path.isfile(bai_target):
os.symlink(hda.metadata.bam_index.file_name, bai_target)
Now the BAM file has a .bai file with the same prefix, and we can run the idxstats
as follows:
## Extract idxstats
import pysam
bam_idxstats_data = pysam.idxstats(hda.file_name)
With the lines of python code above, the idxstats data is parsed into the RAM of python during compilation on the server, but is not yet exported into the HTML page nor parsed by JS. To do that, we add an HTML section to the end of the mako file.
<html>
<head>
<title>${hda.name | h} | ${visualization_name | h}</title>
</head>
<body>
${bam_idxstats_data | h}
</body>
</html>
Here you see ${bam_idxstats_data | h}, which prints the python variable into
the HTML page and also does HTML escaping by providing the ` | h`-flag (for security reasons).
Let’s put this all together.
Hands On: Data upload
- Create the mako file
templates/alignment_rname_boxplot.makoFill it with the following code:
<!DOCTYPE HTML> <% import os ## Generates hash (hdadict['id']) of history item hdadict = trans.security.encode_dict_ids( hda.to_dict() ) ## Finds the parent directory of galaxy (/, /galaxy, etc.) root = h.url_for( '/' ) ## Determines the exposed URL of the ./static directory app_root = root + 'plugins/visualizations/'+visualization_name+'/static/' ## Actual file URL: file_url = os.path.join(root, 'datasets', hdadict['id'], "display?to_ext="+hda.ext) ## Ensure BAI index is symlinked bai_target = hda.file_name+'.bai' if not os.path.isfile(bai_target): os.symlink(hda.metadata.bam_index.file_name, bai_target) ## Extract idxstats import pysam bam_idxstats_data = pysam.idxstats(hda.file_name) %> <html> <head> <title>${hda.name | h} | ${visualization_name | h}</title> </head> <body> ${bam_idxstats_data | h} </body> </html>We are now ready to test this very basic visualization, we just need a (small) BAM file for it.
- Download the example BAM file
Go the galaxy root directory and start Galaxy:
$ cd $GALAXY_ROOT $ ./run.shUpload the example BAM file to your history
If everything went well, our plugin has appeared as a visualization option for the dataset
CommentYou must be logged in to be able to use visualizations
All the visualization does at the moment, is show the contents of idxstats, compiled to HTML:
['chrA\t5386\t2\t0\n', 'chrB\t5386\t4\t0\n', 'chrC\t5386\t1\t0\n', 'chrD\t5386\t6\t1\n',
'chrE\t5386\t3\t0\n', 'chrF\t5386\t2\t0\n', 'chrG\t5386\t1\t0\n', '*\t0\t0\t0\n']
It contains eight entries, one for each of our (made-up) chromosomes and one to *, which represents
the unmapped reads. Entries are tab delimited (\t) and for the chrA entry it indicates that the
length of the RNAME (chromosome) is 5386 bases and 2 reads are aligned to it.
To make the data a bit more usable for Javascript , we convert it into a simple dictionary of the following syntax:
{'chrA':2, 'chrB':4, 'chrC':1, 'chrD':6, 'chrE':3, 'chrF':2, 'chrG':1, '*': 0}
Although it is possible to do this in python we recommend doing this in JS. The var dump provided by python/pysam is actually a valid syntax for Javascript too, so getting the raw data into Javascript is rather easy:
<script>
bam_idxstats_data = ${bam_idxstats_data};
</script>
Converting the data is not the scope of the tutorial, so here we provide such a function:
<script>
bam_idxstats_data = ${bam_idxstats_data};
function parse_data(bam_idxstats_data) {
var output = {};
for(var i = 0; i < data.length ; i++) {
var line = data[i];
var chunks = line.split("\t");
if(chunks[0].split("_").length == 1) { // only if it does not contain underscore
output[chunks[0]] = parseInt(chunks[2]);
}
}
return output;
}
</script>
The great thing about the mako system is that it does not require to restart galaxy in order to make functional changes to the mako files.
Hands On: Data upload
Change the mako file to the following:
<!DOCTYPE HTML> <% import os ## Generates hash (hdadict['id']) of history item hdadict = trans.security.encode_dict_ids( hda.to_dict() ) ## Finds the parent directory of galaxy (/, /galaxy, etc.) root = h.url_for( '/' ) ## Determines the exposed URL of the ./static directory app_root = root + 'plugins/visualizations/'+visualization_name+'/static/' ## Actual file URL: file_url = os.path.join(root, 'datasets', hdadict['id'], "display?to_ext="+hda.ext) ## Ensure BAI index is symlinked bai_target = hda.file_name+'.bai' if not os.path.isfile(bai_target): os.symlink(hda.metadata.bam_index.file_name, bai_target) ## Extract idxstats import pysam bam_idxstats_data = pysam.idxstats(hda.file_name) %> <html> <head> <title>${hda.name | h} | ${visualization_name | h}</title> <script> bam_idxstats_data = ${bam_idxstats_data}; function parse_data(data) { var output = {}; for(var i = 0; i < data.length ; i++) { var line = data[i]; var chunks = line.split("\t"); if(chunks[0].split("_").length == 1) { // only if it does not contain underscore output[chunks[0]] = parseInt(chunks[2]); } } return output; } </script> </head> <body onload="bam_idxstats = parse_data(bam_idxstats_data);"> ${bam_idxstats_data | h} </body> </html>Retrigger the visualization and open the developers console of your browser: In the console, type:
bam_idxstats_dataand press Enter This should give the parsed contents as a dictionary, which can directly be used in Javascript.
From this point forward you are encouraged to continue on your own to see if you are able to create a simple visualization from this dictionary. Think of tables, DIVs or even more complicated solutions :).
Below is an example visualization, which creates a bar plot showing the number of reads per chromosome.
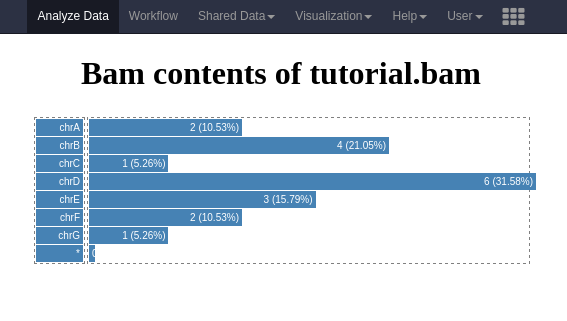
The full contents of this plugin are provided in the GitHub repository related to this material in tree/master/topics/dev/files/hands_on-visualizations/alignment_rname_boxplot.
To try out this example, simply copy this folder to the $GALAXY_ROOT/config/plugins/visualizations/ folder
on your (local) Galaxy and restart Galaxy.
The contents of the mako file for this example are given below.
<!DOCTYPE HTML>
<%
import os
## Generates hash (hdadict['id']) of history item
hdadict = trans.security.encode_dict_ids( hda.to_dict() )
## Finds the parent directory of galaxy (/, /galaxy, etc.)
root = h.url_for( '/' )
## Determines the exposed URL of the ./static directory
app_root = root + 'plugins/visualizations/'+visualization_name+'/static/'
## Actual file URL:
file_url = os.path.join(root, 'datasets', hdadict['id'], "display?to_ext="+hda.ext)
## Ensure BAI index is symlinked
bai_target = hda.file_name+'.bai'
if not os.path.isfile(bai_target):
os.symlink(hda.metadata.bam_index.file_name, bai_target)
## Extract idxstats
import pysam
bam_idxstats_data = pysam.idxstats(hda.file_name)
%>
<html>
<head>
<title>${hda.name | h} | ${visualization_name | h}</title>
<style>
.chart div {
font: 10px sans-serif;
background-color: steelblue;
text-align: right;
padding: 3px;
margin: 1px;
color: white;
}
</style>
<script>
bam_idxstats_data = ${bam_idxstats_data};
function parse_data(data) {
/*
Data comes in as tuple of unsplit lines:
["chr1\t1000\t0\t0", "chr2\t2500\t0\t0"]
We need to split it up, and ideally only keep reference names without an underscore
*/
var output = {};
for(var i = 0; i < data.length ; i++) {
var line = data[i];
var chunks = line.split("\t");
if(chunks[0].split("_").length == 1) { // only if it does not contain underscore
output[chunks[0]] = parseInt(chunks[2]);
}
}
return output;
}
function calc_stats(parsed) {
max = 0;
sum = 0;
for (var key in parsed) {
if (parsed[key] > max){
max = parsed[key];
}
sum += parsed[key]
}
return [max, sum];
}
function plot_data(parsed) {
var max = calc_stats(parsed)[0];
var sum = calc_stats(parsed)[1];
for (var key in parsed) {
var value = parsed[key];
var ratio = 100.0 * value / sum;
var ratio2 = 100.0 * value / max;
var div = document.createElement("div");
div.innerHTML = '<nobr>'+key+'</nobr>';
document.getElementById("chart_names").appendChild(div);
var div = document.createElement("div");
div.innerHTML = '<nobr>'+value+" ("+Math.round(ratio*100)/100+"%)</nobr>";
div.title = key+': '+value+" ("+Math.round(ratio*100)/100+"%)";
div.style.width = ratio2+'%';
document.getElementById("chart").appendChild(div);
}
}
</script>
</head>
<body onload="plot_data(parse_data(bam_idxstats_data));">
<center>
<h1>Bam contents of ${hda.name | h}</h1>
<table border="0" borderpadding="0" borderpanning=")" style="width: 500px;">
<tr>
<td style="width:50px;">
<div id="chart_names" class="chart" style="width: 100%; border: 1px dashed gray;text-align: left;" />
</td>
<td style="width:450px;">
<div id="chart" class="chart" style="width:100%; border: 1px dashed gray;text-align: left;" />
</td>
</tr>
</table>
</center>
</body>
</html>
In the given example, the RNAME queries containing an underscore were removed.
This is because there are many alternative chromosomes, making the list very large
for certain reference genomes. However, for certain studies it might be desired to
just look at those.
The current plot is a box plot, but one can imagine a pie-chart may be convenient too. All of those additional settings can be implemented for interactive behaviour, contributing to quicker understanding of the data which is generally not so convenient using static Galaxy tools.
Comment: Static filesIn the example we included Javascript and CSS into the HTML website. Remember that for every new invocation of the visualization the entire CSS en JS are copied and transferred as well. This is a waste of (redundant) bandwidth as we could save the files in the static directory and refer to them within the HTML. The browser shall check it’s cache for the presence of libs and style sheets and only update them if they have changed.
Improvements
Another thing you may realize is that we still do the calculation (pysam.idxstats) server side. Although this is a marginal calculation, it is causing delay and the bigger the files, the worse this delay becomes. The underlying problem is that we did not obtain and parse the BAI file via Javascript. This would be a more elaborate solution, but requires more development time as we need to develop a function able to parse the binary file.
Another thing we could do is create a static samtools idxstats tool, that creates a file of
datatype tabular.Idxstats and include that datatype into Galaxy. We then make the visualization
specific for that datatype, just plotting the results of the idxstats.
A fundamental and more complicated problem is that BAM files are simply too big to transfer for these kind of applications. It would be ideal to have web server integration that allows querying of specific locations or metadata within or from a BAM file where indexing operations are taken care of at the server side. This is what has been done in Trackster.
More examples
For more examples of visualization plugins, you can browse this GitHub repo
Conclusion
We have just created a visualization plugin in Galaxy to visualize the number of alignments
per RNAME (chromosome) in a BAM file.
You've Finished the Tutorial
Key points
Visualizations require a different way of thinking: server and client side; downloading files rather than system level access
Interactivity is what makes visualizations different from static tools
Requires understanding of both the Galaxy ecosystem as well as HTML5/JS
Performance is more important than for static Galaxy tools
Frequently Asked Questions
Have questions about this tutorial? Have a look at the available FAQ pages and support channelsFeedback
Did you use this material as an instructor? Feel free to give us feedback on how it went.
Did you use this material as a learner or student? Click the form below to leave feedback.

Citing this Tutorial
- Saskia Hiltemann, Youri Hoogstrate, Generic plugins (Galaxy Training Materials). https://training.galaxyproject.org/training-material/topics/dev/tutorials/visualization-generic/tutorial.html Online; accessed TODAY
- Hiltemann, Saskia, Rasche, Helena et al., 2023 Galaxy Training: A Powerful Framework for Teaching! PLOS Computational Biology 10.1371/journal.pcbi.1010752
- Batut et al., 2018 Community-Driven Data Analysis Training for Biology Cell Systems 10.1016/j.cels.2018.05.012
Congratulations on successfully completing this tutorial!@misc{dev-visualization-generic, author = "Saskia Hiltemann and Youri Hoogstrate", title = "Generic plugins (Galaxy Training Materials)", year = "", month = "", day = "", url = "\url{https://training.galaxyproject.org/training-material/topics/dev/tutorials/visualization-generic/tutorial.html}", note = "[Online; accessed TODAY]" } @article{Hiltemann_2023, doi = {10.1371/journal.pcbi.1010752}, url = {https://doi.org/10.1371%2Fjournal.pcbi.1010752}, year = 2023, month = {jan}, publisher = {Public Library of Science ({PLoS})}, volume = {19}, number = {1}, pages = {e1010752}, author = {Saskia Hiltemann and Helena Rasche and Simon Gladman and Hans-Rudolf Hotz and Delphine Larivi{\`{e}}re and Daniel Blankenberg and Pratik D. Jagtap and Thomas Wollmann and Anthony Bretaudeau and Nadia Gou{\'{e}} and Timothy J. Griffin and Coline Royaux and Yvan Le Bras and Subina Mehta and Anna Syme and Frederik Coppens and Bert Droesbeke and Nicola Soranzo and Wendi Bacon and Fotis Psomopoulos and Crist{\'{o}}bal Gallardo-Alba and John Davis and Melanie Christine Föll and Matthias Fahrner and Maria A. Doyle and Beatriz Serrano-Solano and Anne Claire Fouilloux and Peter van Heusden and Wolfgang Maier and Dave Clements and Florian Heyl and Björn Grüning and B{\'{e}}r{\'{e}}nice Batut and}, editor = {Francis Ouellette}, title = {Galaxy Training: A powerful framework for teaching!}, journal = {PLoS Comput Biol} }