Running molecular dynamics simulations using NAMD
Under Development!
This tutorial is not in its final state. The content may change a lot in the next months. Because of this status, it is also not listed in the topic pages.
| Author(s) |
|
| Reviewers |
|
OverviewQuestions:
Objectives:
How do I use the NAMD engine in Galaxy?
What is the correct procedure for performing a simple molecular dynamics simulation of a protein?
Requirements:
Learn about the NAMD engine provided in BRIDGE
Time estimation: 3 hoursLevel: Intermediate IntermediateSupporting Materials:Published: Jun 3, 2019Last modification: Nov 9, 2023License: Tutorial Content is licensed under Creative Commons Attribution 4.0 International License. The GTN Framework is licensed under MITpurl PURL: https://gxy.io/GTN:T00052version Revision: 9
In this tutorial we will perform a simulation with the popular NAMD molecular dynamics software. Please note NAMD tools are not currently available on a public Galaxy server due to licensing issues. If you are interested in following this tutorial, you will need to download the BRIDGE docker container and download NAMD yourself.
AgendaIn this tutorial, we will cover:
This tutorial is made up of two parts. In the first section, we will look at preparation of a system (solvation, charge neutralisation, energy minimisation) using CHARMM. In the second section, we will perform an equilibration and production simulation, using NAMD. If you already completed the Setting up molecular systems tutorial, which covers the use of the CHARMM graphical user interface (GUI), you have already prepared your system, so go straight to the second section, using the files you prepared earlier.
The process can be accomplished by selecting each tool from the tools menu, or by importing the workflow. The workflow method is most efficient and the individual tools used in the workflows are discussed below. The entire workflow (preparation + simulation) is shown below for CHARMM and NAMD.
See Statistical Mechanics, McQuarrie for more in depth theory ISBN:9781891389153
System preparation with CHARMM
If you already prepared your system using the CHARMM-GUI, and saved the output files, you can skip this section.
Setup
Initially, we need to prepare a protein-ligand system in CHARMM.
This tool will:
- solvate the protein-ligand complex, using the TIP3P water model
- neutralise the system, using 0.05M NaCl
- conduct a short energy minimisation
Hands On: Data upload
Create a new history for this tutorial.
To create a new history simply click the new-history icon at the top of the history panel:
- Import the files from the Zenodo link provided.
https://zenodo.org/record/3234841/files/cbh1test.crd https://zenodo.org/record/3234841/files/cbh1test.psf
- Copy the link location
Click galaxy-upload Upload at the top of the activity panel
- Select galaxy-wf-edit Paste/Fetch Data
Paste the link(s) into the text field
Press Start
- Close the window
As an alternative to uploading the data from a URL or your computer, the files may also have been made available from a shared data library:
- Go into Libraries (left panel)
- Navigate to the correct folder as indicated by your instructor.
- On most Galaxies tutorial data will be provided in a folder named GTN - Material –> Topic Name -> Tutorial Name.
- Select the desired files
- Click on Add to History galaxy-dropdown near the top and select as Datasets from the dropdown menu
In the pop-up window, choose
- “Select history”: the history you want to import the data to (or create a new one)
- Click on Import
Rename the datasets.
- Click on the galaxy-pencil pencil icon for the dataset to edit its attributes
- In the central panel, change the Name field
- Click the Save button
Check that the datatype is correct. The crd file should have the CRD datatype and the psf file the PSF datatype.
- Click on the galaxy-pencil pencil icon for the dataset to edit its attributes
- In the central panel, click galaxy-chart-select-data Datatypes tab on the top
- In the galaxy-chart-select-data Assign Datatype, select
datatypesfrom “New Type” dropdown
- Tip: you can start typing the datatype into the field to filter the dropdown menu
- Click the Save button


Hands On: initial processingRun System Setup tool with the following parameters:
- param-file “psf input”: Protein structure file of the protein-ligand system. (Input dataset)
- param-file “crd input”: Coordinate file of the protein-ligand system. (Input dataset)
- “Buffer”: Edge to edge distance between the protein and the edge of the water box in angstroms
- “Custom topology and parameter files for ligands” (optional)
Energy minimisation
The setup provides us with the CRD and PSF files needed to perform a simulation. In addition, we have a PRM file which defines the parameters of the unit cell. Now, we need to perform energy minimisation. This relaxes the system, removing any steric clashes or unusual geometry which could artificially raise the energy.
This tools will:
- Minimise energy using a steepest descent algorithm followed by Adopted Newton Raphson (using the defined number of steps)
- Set up periodic boundaries and generate Particle Mesh Ewald (PME)
- generate reference structures for restraints in NAMD (if selected)
Hands On: energy minimizationRun Energy Minimizer tool with the following parameters:
- param-file “system_setup_crd”: Coordinate file generated by the setup tool. (Input dataset)
- param-file “system_setup_psf”: Protein structure file generated by the setup tool. (Input dataset)
- param-file “waterbox parameters input”: Water box parameter file generated by the setup tool. (Input dataset)
- “Minimization steps”:
1000- “Create reference structures for RMSD restraints for NAMD?”:
No- “Custom topology and parameter files for ligands”:
No
MD simulations with NAMD
At this point we are ready to run the simulation, which uses NAMD as a molecular dynamics engine. An NVT simulation is followed by an NPT simulation.
NVT
Classical NVT dynamics, maintaining constant number of particles, volume and temperature.
This tool runs classical molecular dynamics simulations in NAMD using an NVT ensemble. User can run the simulation in small time intervals. The coordinates, velocities and the extended system files can be use to restart the simulations. If required, harmonic restraints can be used to maintain the protein shape. These restraints, in particular RMSD harmonic restraints can be added with the NAMD collective variable module.
Hands On: NVT dynamics
- NAMD MD Simulator (NVT) tool with the following parameters:
- param-file “xplor psf input”: PSF file from CHARMM preparation (Input dataset)
- param-file “pdb input”: PDB file from CHARMM preparation (Input dataset)
- param-file “PME grid specs”: Generated by the setup tool (Input dataset)
- param-file “waterbox prm input”: Water box parameter file generated by the setup tool. (Input dataset)
- “Temperature (K)”:
300- “Are you restarting a simulation?”:
No- “Use Harmonic restraints simulation?”:
No- “Custom parameter file for ligands”:
No- “DCD Frequency (ps)”:
1(Frequency to record frames in the DCD trajectory)- “Simulation Time (ps)”:
10- “Number of processors”:
4
NPT
Classical NPT dynamics, maintaining constant number of particles, pressure and temperature.
This tool runs classical molecular dynamics simulations in NAMD using an NPT ensemble. User can run the simulation in small time intervals. The coordinates, velocities and the extended system files can be use to restart the simulations. Harmonic restraints can be used. NAMD collective variable module is used to give RMSD harmonic restraints.
Hands On: NPT dynamics
- NAMD MD Simulator (NPT) tool with the following parameters:
- param-file “xplor psf input”: PSF file from CHARMM preparation (Input dataset)
- param-file “pdb input”: PDB file from CHARMM preparation (Input dataset)
- param-file “PME grid specs”: Generated by the setup tool (Input dataset)
- param-file “waterbox prm input”: Water box parameter file generated by the setup tool. (Input dataset)
- “Temperature (K)”:
300- “Pressure (bar)”:
1.01325- “Are you restarting a simulation?”:
Yes- “Coordinates from the previous simulation”: from the NVT simulation
- “Velocities from the previous simulation”: from the NVT simulation
- “Extended system of the previous simulation”: from the NVT simulation
- “Use Harmonic restraints simulation?”:
No- “Custom parameter file for ligands”:
No- “DCD Frequency (ps)”:
1(Frequency to record frames in the DCD trajectory)- “Simulation Time (ps)”:
15- “Number of processors”:
4
Workflows
Both the CHARMM preparatory workflow and the NAMD simulation workflow are available as an alternative to executing individual tools.
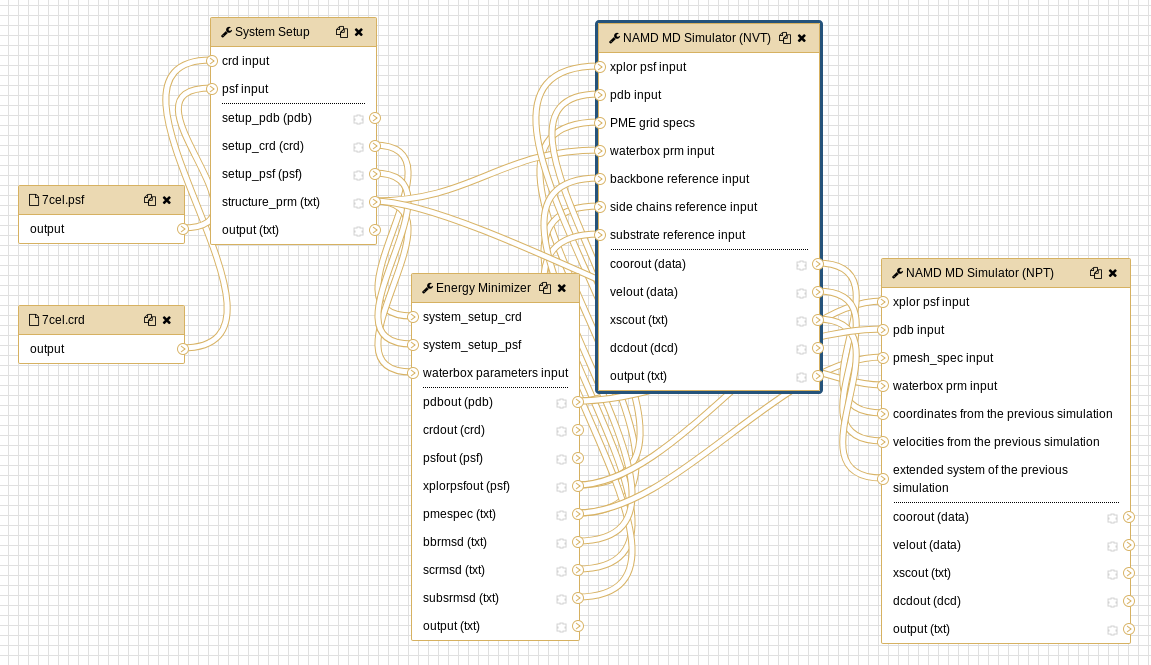 Open image in new tab
Open image in new tab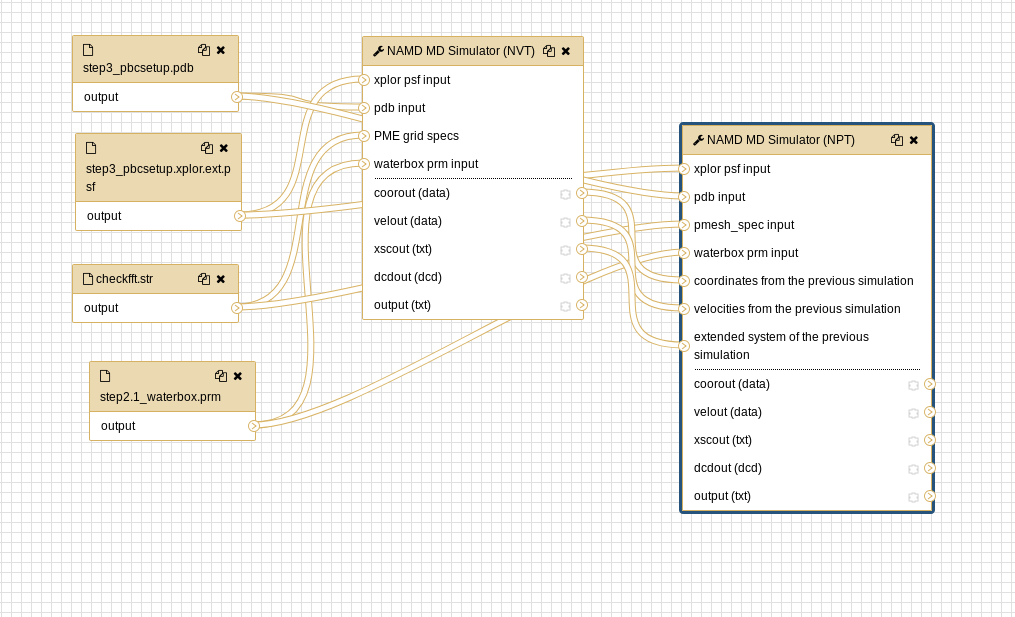 Open image in new tab
Open image in new tabConclusion
After completing the steps, or running the workflow, we have successfully produced a trajectory (the xtc file) which describes the atomic motion of the system. This can be viewed using molecular visualization software or analysed further; please visit the visualization and analysis tutorials for more information.
You've Finished the Tutorial
Key points
Several MD engines are available in BRIDGE
Workflows are available for common simulations tasks such as equilibration and production dynamics for various ensembles (NVT, NPT)
You’ve run an equilibrium and production MD simulation using NAMD
Frequently Asked Questions
Have questions about this tutorial? Have a look at the available FAQ pages and support channelsUseful literature
Further information, including links to documentation and original publications, regarding the tools, analysis techniques and the interpretation of results described in this tutorial can be found here.
Feedback
Did you use this material as an instructor? Feel free to give us feedback on how it went.
Did you use this material as a learner or student? Click the form below to leave feedback.

Citing this Tutorial
- Christopher Barnett, Tharindu Senapathi, Simon Bray, Running molecular dynamics simulations using NAMD (Galaxy Training Materials). https://training.galaxyproject.org/training-material/topics/computational-chemistry/tutorials/md-simulation-namd/tutorial.html Online; accessed TODAY
- Hiltemann, Saskia, Rasche, Helena et al., 2023 Galaxy Training: A Powerful Framework for Teaching! PLOS Computational Biology 10.1371/journal.pcbi.1010752
- Batut et al., 2018 Community-Driven Data Analysis Training for Biology Cell Systems 10.1016/j.cels.2018.05.012
Congratulations on successfully completing this tutorial!@misc{computational-chemistry-md-simulation-namd, author = "Christopher Barnett and Tharindu Senapathi and Simon Bray", title = "Running molecular dynamics simulations using NAMD (Galaxy Training Materials)", year = "", month = "", day = "", url = "\url{https://training.galaxyproject.org/training-material/topics/computational-chemistry/tutorials/md-simulation-namd/tutorial.html}", note = "[Online; accessed TODAY]" } @article{Hiltemann_2023, doi = {10.1371/journal.pcbi.1010752}, url = {https://doi.org/10.1371%2Fjournal.pcbi.1010752}, year = 2023, month = {jan}, publisher = {Public Library of Science ({PLoS})}, volume = {19}, number = {1}, pages = {e1010752}, author = {Saskia Hiltemann and Helena Rasche and Simon Gladman and Hans-Rudolf Hotz and Delphine Larivi{\`{e}}re and Daniel Blankenberg and Pratik D. Jagtap and Thomas Wollmann and Anthony Bretaudeau and Nadia Gou{\'{e}} and Timothy J. Griffin and Coline Royaux and Yvan Le Bras and Subina Mehta and Anna Syme and Frederik Coppens and Bert Droesbeke and Nicola Soranzo and Wendi Bacon and Fotis Psomopoulos and Crist{\'{o}}bal Gallardo-Alba and John Davis and Melanie Christine Föll and Matthias Fahrner and Maria A. Doyle and Beatriz Serrano-Solano and Anne Claire Fouilloux and Peter van Heusden and Wolfgang Maier and Dave Clements and Florian Heyl and Björn Grüning and B{\'{e}}r{\'{e}}nice Batut and}, editor = {Francis Ouellette}, title = {Galaxy Training: A powerful framework for teaching!}, journal = {PLoS Comput Biol} }
Do you want to extend your knowledge?Follow one of our recommended follow-up trainings:
You can use Ephemeris's
shed-tools installcommand to install the tools used in this tutorial.shed-tools install [-g GALAXY] [-a API_KEY] -t <(curl https://training.galaxyproject.org/training-material/api/topics/computational-chemistry/tutorials/md-simulation-namd/tutorial.json | jq .admin_install_yaml -r)Alternatively you can copy and paste the following YAML
--- install_tool_dependencies: true install_repository_dependencies: true install_resolver_dependencies: true tools: []