name: inverse layout: true class: center, middle, inverse <div class="my-header"><span> <a href="/training-material/topics/single-cell" title="Return to topic page" ><i class="fa fa-level-up" aria-hidden="true"></i></a> <a href="https://github.com/galaxyproject/training-material/edit/main/topics/single-cell/tutorials/scrna-plates-batches-barcodes/slides.html"><i class="fa fa-pencil" aria-hidden="true"></i></a> </span></div> <div class="my-footer"><span> <img src="/training-material/assets/images/GTN-60px.png" alt="Galaxy Training Network" style="height: 40px;"/> </span></div> --- <img src="/training-material/assets/images/GTNLogo1000.png" alt="Galaxy Training Network" class="cover-logo"/> <br/> <br/> # Plates, Batches, and Barcodes <br/> <br/> <div markdown="0"> <div class="contributors-line"> <ul class="text-list"> <li> <a href="/training-material/hall-of-fame/mtekman/" class="contributor-badge contributor-mtekman"><img src="https://avatars.githubusercontent.com/mtekman?s=36" alt="Mehmet Tekman avatar" width="36" class="avatar" /> Mehmet Tekman</a> <li> <a href="/training-material/hall-of-fame/astrovsky01/" class="contributor-badge contributor-astrovsky01"><img src="/training-material/assets/images/orcid.png" alt="orcid logo" width="36" height="36"/><img src="https://avatars.githubusercontent.com/astrovsky01?s=36" alt="Alex Ostrovsky avatar" width="36" class="avatar" /> Alex Ostrovsky</a></li> </ul> </div> </div> <!-- modified date --> <div class="footnote" style="bottom: 8em;"> <i class="far fa-calendar" aria-hidden="true"></i><span class="visually-hidden">last_modification</span> Updated: <i class="fas fa-fingerprint" aria-hidden="true"></i><span class="visually-hidden">purl</span><abbr title="Persistent URL">PURL</abbr>: <a href="https://gxy.io/GTN:S00085">gxy.io/GTN:S00085</a> </div> <!-- other slide formats (video and plain-text) --> <div class="footnote" style="bottom: 5em;"> <i class="far fa-play-circle" aria-hidden="true"></i><span class="visually-hidden">video-slides</span> <a href="/training-material/videos/watch.html?v=/single-cell/tutorials/scrna-plates-batches-barcodes/slides">Video slides</a> | <i class="fas fa-file-alt" aria-hidden="true"></i><span class="visually-hidden">text-document</span><a href="slides-plain.html"> Plain-text slides</a> | </div> <!-- usage tips --> <div class="footnote" style="bottom: 2em;"> <strong>Tip: </strong>press <kbd>P</kbd> to view the presenter notes | <i class="fa fa-arrows" aria-hidden="true"></i><span class="visually-hidden">arrow-keys</span> Use arrow keys to move between slides </div> ??? Presenter notes contain extra information which might be useful if you intend to use these slides for teaching. Press `P` again to switch presenter notes off Press `C` to create a new window where the same presentation will be displayed. This window is linked to the main window. Changing slides on one will cause the slide to change on the other. Useful when presenting. --- ## Requirements Before diving into this slide deck, we recommend you to have a look at: - [Introduction to Galaxy Analyses](/training-material/topics/introduction) - [Single Cell](/training-material/topics/single-cell) - Understanding Barcodes: [<i class="fas fa-laptop" aria-hidden="true"></i><span class="visually-hidden">tutorial</span> hands-on](/training-material/topics/single-cell/tutorials/scrna-umis/tutorial.html) --- ### <i class="far fa-question-circle" aria-hidden="true"></i><span class="visually-hidden">question</span> Questions - What is a batch when it comes to single cells? - What’s the difference between a barcode and an index? - What is batch effect and how do you prevent it? - What is a lane? --- ### <i class="fas fa-bullseye" aria-hidden="true"></i><span class="visually-hidden">objectives</span> Objectives - How to set up plates to prevent batch effect - Proper naming conventions when dealing with scRNA-seq samples --- ### Sorting Plates .image-100[] .left[Plates are *N x M* arrays of wells that cells are sorted, to then be individually amplified and sequenced.] ??? - Sorting plates are 2 dimensional arrays of wells that individual cells are placed into. - Each cell in each well is tagged with a unique barcode and then primed to then be amplified and sequenced - The method in which this is performed can yield significant pros and cons as we will see. --- ### Sorting Plates .image-80[] ??? - Here we see 3 different sorting strategies with different advantages for same treatment sample. - In the top image: We see all wells are filled, to maximise the number of cells on a plate - In the middle image: We see only the inner wells are filled, to maintain a protective border during library preparation - In the bottom image: We see a striping pattern, which mitigates contamination of material from adjoining wells --- ### Setting up Plates .image-75[] .center[What is the problem with this plate setup?] ??? - However, if we consider different treatment samples, then we must consider different challenges. - What is the advantage of the top example, i.e. Several different treatments on one plate? - What is the advantage of the bottom example, i.e. One treatment on one plate? --- ### Setting up Plates .image-50[] .center[Batch effect (plate vs plate) cannot be separated from treatment effect in either scenario.] ??? - Trick question! - In both examples, the treatment effect cannot be separated from any technical effect from the plate itself. - For example, the biological effect of the yellow cells might just be the technical confounding effect from Batch 1. - What can we do instead to reduce the effect of these technical confounding effects across treatments? --- ### Setting up Plates .image-50[] .left[Either of these are better set-ups. Mixing columns is good, but not required. Ultimately, batch effect can now be separated from variable effect.] ??? - The answer is that we can balance the treatments across our plates better. - By ensuring the same treatment occurs in comparable amounts in different plates, we can reduce the effect that each plate would have on the treatment. --- ### Setting up Plates .image-90[] .center[Can't mix samples on plates? Separate replicates evenly and process together.] ??? - If putting multiple treatments on the same plate is not an option, then having enough separated replicates will also allow for batch correction. - Here, you can assess the variation between replicates, and thus batches, as well as between treatments. --- ### What about sequencing lanes? .center[So now it's time to sequence our samples! How do we combine samples into sequencing lanes?] .image-90[] .center[This works well, but what if you have too many samples for one lane?] ??? - So now it's time to sequence our samples! How do we combine samples into sequencing lanes? - In many cases, it is enough to sequence 1 or 2 plates together in a single sequencing lane. - But what if we wish to sequence 8 plates? --- ### What about sequencing lanes? .center[So now it's time to sequence our samples! How do we combine samples into sequencing lanes?] .image-90[] .center[Does this look ok?] ??? - In the example show, Treatment A would go to the first sequencing lane. - Treatment B would go to the second sequencing lane. - Is it okay to sequence different treatments across different sequencing lanes? -- .center[No! You've turned each treatment (A & B) into a batch!] ??? - No, because you've turned Treatment A and Treatment B into separate batches. - This gives us the same unbalanced design we saw with the sorting plates. --- ### What about sequencing lanes? .image-75[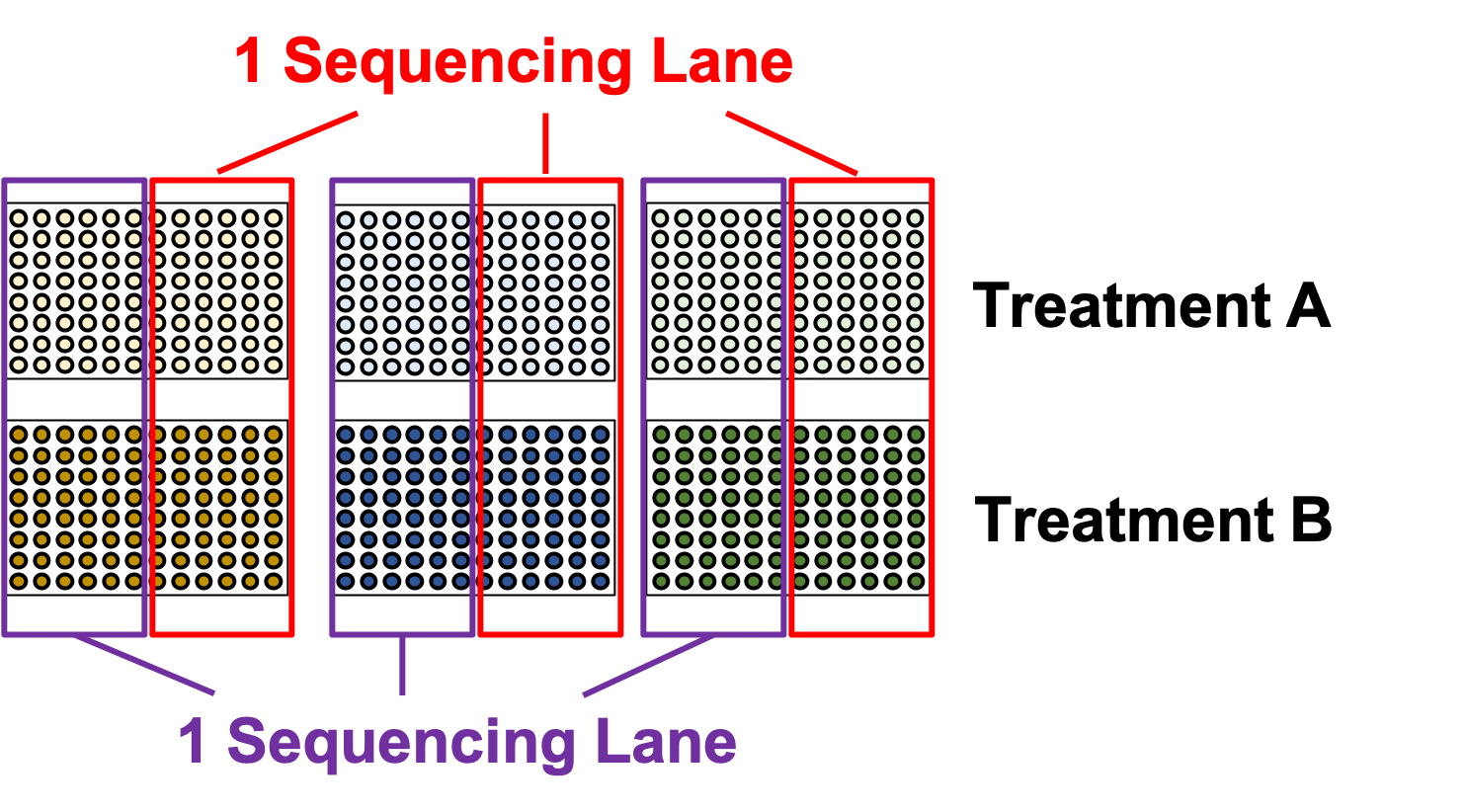] .center[This is the way to balance your batches at the lane-level.] ??? - Instead we should balance the availability of each treatment across different sequencing lanes - This way, each treatment is sequenced at the same time as another treatment, reducing the technical effect from the sequencing on the treatment. --- ### Distinguishing cells in a plate .image-60[] * Cells are selected from a plate by their *barcodes* * Barcodes must be unique + e.g. 96 wells in a plate, need 96 barcodes to sequence them together ??? - How do we distinguish each cell in each well? - Do we remember the physical location of that cell on that well, or do we tag it somehow? - The answer is that each cell is given a unique barcode that distinguishes from other cells which have different cell barcodes. --- ### What are Cell Barcodes? .image-100[] * Barcodes are added to *all* transcripts of a *specific* cell * Transcripts with different cell barcodes originate from different cells ??? - What exactly are cell barcodes? - These are usually short nucleotide barcodes that are added to all transcripts of a specific cell - The idea is that transcripts with different cell barcodes originate from different cells. --- ### What are Cell Barcodes? .image-100[] * Many different cell barcodes are used across many different cells * Each well in a plate contains a cell, indexed by its cell barcode ??? - Within a plate, many different barcodes are used, and each cell is indexed by the cell barcode assigned to that well. --- ### Questions about Cell Barcodes .image-50[] .left[Assuming you sequence one 96-well plate:] 1. How many cell barcodes are needed for a single lane? 1. How many cell barcodes are needed if you combine 10 plates into a single sequencing lane? 1. What would be the minimum length of the barcodes for each of the previous questions? ??? - Consider a 12 by 8 plate with 96 wells. - How many cell barcodes are needed if you sequence a single plate? - How many cell barcodes are needed if you sequence 10 plates into a single sequencing lane? - What would be the minimum length of the barcodes for each of the previous questions? -- .footnote[ 1. *96 unique barcodes per lane* 1. *96 x 10 = 960 unique barcodes per plate* ] ??? - For one plate you would just need 96 unique barcodes - For 10 plates you would need 960 unique barcodes --- ### Questions about Cell Barcodes .left[<small>3) What would be the minimum length of the barcodes for each of the previous questions?</small>] .pull-left[ A single lane? * 96 barcodes, 4 nucleotides for each base of a barcode | Barcodes | Result | |---------|------| | $$4^2 = 16$$ | <small>No, 2 bases is not enough | | $$4^3 = 64$$ | <small>No, 3 bases is not enough | | $$4^4 = 256$$ | <small>Yes, 4 bases is enough to cover 96 barcodes (and more!) | ] ??? - To answer the third question, we need to make some assumptions about the barcodes. - If we assume a barcode is made up of a chain of 4 possible nucleotides, then we can solve the problem as a power of 4. - For a single lane with only 96 barcodes, we require our barcodes to be of at least 4 base pairs in length. -- .pull-right[ 10 plates in a single lane? * 960 barcodes, 4 nucleotides for each base of a barcode | Barcodes | Result | |---------|------| |$$4^4 = 256$$ | <small>No, 4 bases is not enough | |$$4^5 = 1024$$ | <small>Yes, 5 bases is enough to cover 960 barcodes (just barely!) | ] ??? - For 10 lanes with 960 barcodes, we require our barcodes to be of at least 5 base pairs in length. - However these calculations also make one serious assumption about the barcodes. --- ### Barcode Safeguarding .pull-left[ * Is 5 nucleotides really enough to capture 960 cells? * What could go wrong if all barcodes are separated by 1 bp, as shown? ] .pull-right[ <small>*Edit distance = 1bp* AAAAA AAAAC AAAAG AAAAT AAACA AAAGA AAATA ···· CCCCC CCCCA CCCCG CCCCT CCCAC CCCGC CCCTC ···· · · · ] ??? - For example, we assume that all our barcodes are at minimum separated by a single base pair. - This is known as an edit distance of 1. - If we assume that every barcode is separated from every other barcode by 1 base pair, then 5 nucleotides is indeed enough to delineate 960 cells. - However if there was even just one small sequencing error of 1 base pair on any barcode, it would immediately change that erronous barcode into another real barcode. - That is to say, we have no way of detecting sequencing errors if we use an edit distance of 1. --- ### Guarding against Sequencing Errors .pull-left[ * Sequencing errors can change the cell barcode * To guard against a 1 bp sequencing error, need to space barcodes by 2 bp * How many barcodes for a cell barcode of length 5 bp (ED = 2)? ] .pull-right[ <small>*Edit Distance = 2 bp* AAAAA AAACC AAAGG AAATT AACCA AAGGA AATTA ···· CCCCC CCCAA CCCGG CCCTT CCCAA CCGGC CCTTC ···· · · · ] ??? - Let us try to rectify this by increasing the edit distance to 2. - In the example barcodes shown we see barcodes of length 5, separated by 2 base pairs. - How many unique cell barcodes can we make given these restrictions? -- $$ 4^{5-1} = 512 $$ ??? - The answer is half the amount that we need. --- ### Edit Distance : General Principle .pull-left[ e.g. For barcodes of length N=3: * Edit Distance of E=1: .center[`AAA AAC AAG AAT ACA ACC` ...] .center[<small>64 barcodes</small>] * Edit Distance of E=2: .center[`AAA ACC AGG ATT CAA CCC` ...] .center[<small>16 barcodes</small>] * Edit Distance of E=3: .center[`AAA CCC GGG TTT`] .center[<small>4 barcodes</small>] ] ??? - Let us explore this more explicitly. - Here we will be using barcodes only of length 3, and we will explore different edit distances of 1, 2, and 3. - These yield 64, 16, and 4 barcodes respectively. -- .pull-right[ Number of Barcodes : $$ 4^{N-(E-1)} $$ .center[For barcodes of length *N*, and Edit Distance of *E*.] ] ??? - This can be summarized by the formula given, where the length of the barcodes, minus one minus the edit distance, gives the number of available barcodes. --- ### How many available barcodes are there? .pull-left[ * Barcodes typically limited to 4 main bases {A,C,T,G}. * Number of available barcodes depends on: 1. The length of the barcode 2. The edit distance between adjacent barcodes * Must take sequencing errors into account ] ??? - To summarize, barcodes are typically limited to 4 nucleotides, and the number of available barcodes depends on the length and edit distance. - This is to ensure that sequencing errors are taken into account. -- .pull-right[ * Availability is balance between *barcode size* vs. *sequencing errors* * Typically between 4-10 bases, range of cells that can be labelled: $$[4^4, 4^{10}] = [256, 1048576]$$ * True range is lower due to edit distance. ] ??? - The availability of barcodes and the number of cells they can label is a design choice, where the technician must balance two opposing forces: barcode size vs sequencing errors. - For the size, this means that for a barcode N bases long, there will 4 to the N barcodes available. - Typically barcodes tend to span 4 to 10 bases, since longer barcodes tend to be more subjectable to sequencing errors. - The true number of barcodes used is smaller than 4 to the N, due to the countermeasures used to space barcodes apart from one another, in order to reduce sequencing errors. --- ### Design Factors in Barcodes Is it better to use Longer or Shorter Barcodes? | Longer | Shorter | |--------|---------| | Offer larger range of barcodes | Offer smaller range of barcodes | | Prone to more sequencing errors | More robust against sequencing errors | | Must increase edit distance significantly | A smaller edit distance is more acceptable | | Can accommodate large edit distances | *Cannot* accommodate large edit distances | ??? - To weigh the pros and cons of longer and shorter barcodes, we need to take into account the likelihood of sequencing errors for a given size, and the resulting barcode address space given by the edit distance. --- ### Cell Barcodes: Summary .pull-left[ * A single barcode sequence indexes a single cell * Every transcript in a specific cell has the same cell barcode * Barcodes are *designed* for smaller plate-based protocols, while for split-pool and similar techniques they are randomised. * Barcode use is *limited* by length and read depth ] ??? - To summarize cell barcodes, we should note the following - A single barcode sequence indexes a single cell - Every transcript in a specific cell has the same cell barcode - Barcodes are designed for smaller plate-based protocols, while for split-pool and similar techniques they are randomised - Barcode use is limited by length and read depth -- <hline> .pull-right[#### Need to balance: <small> | | | | |-|-|-| | Edit Distance | vs. | Barcode Length | | Number of Barcodes | vs. | Plate and Lane Size | </small> ] ??? - The number of reads wanted per cell determines how many cells you run in a sequencing lane, which in turn tells you how many barcodes you need. --- # Summary .pull-left[ * Cell Barcodes are sequences attached to transcripts to indicate what cell a transcript came from * Cell Barcodes are *designed* to the Plate/Lane setup ] ??? - From the content shown here, you have learned the following. - Cell Barcodes are sequences attached to transcripts to indicate what cell a transcript came from - Cell Barcodes are designed for the Plate and Lane setup. -- .pull-right[ * Indicates what cell a transcript came from. * Reduce sequencing errors ] ??? - Cell barcodes indicate what cell a transcript came from - They reduce sequencing errors when spaced appropriately --- ### <i class="fas fa-key" aria-hidden="true"></i><span class="visually-hidden">keypoints</span> Key points - Balanced batches and replicates allow bioinformatic batch correction - A sequencing lane often contains multiple batches, and is itself a batch effect! --- ### <i class="fas fa-graduation-cap" aria-hidden="true"></i><span class="visually-hidden">curriculum</span> Do you want to extend your knowledge? Follow one of our recommended follow-up trainings: - [Single Cell](/training-material/topics/single-cell) - Pre-processing of 10X Single-Cell RNA Datasets: [<i class="fas fa-laptop" aria-hidden="true"></i><span class="visually-hidden">tutorial</span> hands-on](/training-material/topics/single-cell/tutorials/scrna-preprocessing-tenx/tutorial.html) --- ## Thank You! This material is the result of a collaborative work. Thanks to the [Galaxy Training Network](https://training.galaxyproject.org) and all the contributors! <div markdown="0"> <div class="contributors-line"> <table class="contributions"> <tr> <td><abbr title="These people wrote the bulk of the tutorial, they may have done the analysis, built the workflow, and wrote the text themselves.">Author(s)</abbr></td> <td> <a href="/training-material/hall-of-fame/mtekman/" class="contributor-badge contributor-mtekman"><img src="https://avatars.githubusercontent.com/mtekman?s=36" alt="Mehmet Tekman avatar" width="36" class="avatar" /> Mehmet Tekman</a><a href="/training-material/hall-of-fame/astrovsky01/" class="contributor-badge contributor-astrovsky01"><img src="/training-material/assets/images/orcid.png" alt="orcid logo" width="36" height="36"/><img src="https://avatars.githubusercontent.com/astrovsky01?s=36" alt="Alex Ostrovsky avatar" width="36" class="avatar" /> Alex Ostrovsky</a> </td> </tr> <tr> <td><abbr title="These people edited the text, either for spelling and grammar, flow, GTN-fit, or other similar editing categories">Editor(s)</abbr></td> <td> <a href="/training-material/hall-of-fame/nomadscientist/" class="contributor-badge contributor-nomadscientist"><img src="/training-material/assets/images/orcid.png" alt="orcid logo" width="36" height="36"/><img src="https://avatars.githubusercontent.com/nomadscientist?s=36" alt="Wendi Bacon avatar" width="36" class="avatar" /> Wendi Bacon</a></td> </tr> <tr class="reviewers"> <td><abbr title="These people reviewed this material for accuracy and correctness">Reviewers</abbr></td> <td> <a href="/training-material/hall-of-fame/hexylena/" class="contributor-badge contributor-badge-small contributor-hexylena"><img src="https://avatars.githubusercontent.com/hexylena?s=36" alt="Helena Rasche avatar" width="36" class="avatar" /></a><a href="/training-material/hall-of-fame/bgruening/" class="contributor-badge contributor-badge-small contributor-bgruening"><img src="https://avatars.githubusercontent.com/bgruening?s=36" alt="Björn Grüning avatar" width="36" class="avatar" /></a><a href="/training-material/hall-of-fame/shiltemann/" class="contributor-badge contributor-badge-small contributor-shiltemann"><img src="https://avatars.githubusercontent.com/shiltemann?s=36" alt="Saskia Hiltemann avatar" width="36" class="avatar" /></a><a href="/training-material/hall-of-fame/mtekman/" class="contributor-badge contributor-badge-small contributor-mtekman"><img src="https://avatars.githubusercontent.com/mtekman?s=36" alt="Mehmet Tekman avatar" width="36" class="avatar" /></a><a href="/training-material/hall-of-fame/nomadscientist/" class="contributor-badge contributor-badge-small contributor-nomadscientist"><img src="https://avatars.githubusercontent.com/nomadscientist?s=36" alt="Wendi Bacon avatar" width="36" class="avatar" /></a></td> </tr> </table> </div> </div> <div style="display: flex;flex-direction: row;align-items: center;justify-content: center;"> <img src="/training-material/assets/images/GTNLogo1000.png" alt="Galaxy Training Network" style="height: 100px;"/> </div> Tutorial Content is licensed under <a rel="license" href="http://creativecommons.org/licenses/by/4.0/">Creative Commons Attribution 4.0 International License</a>.<br/>