Metaproteomics is the large-scale characterization of the entire complement of proteins expressed by microbiota. However, metaproteomics analysis of clinical samples is challenged by the presence of abundant human (host) proteins which hampers the confident detection of lower abundant microbial proteins Batut et al. 2018 ; Jagtap et al. 2015 .
To address this, we used tandem mass spectrometry (MS/MS) and bioinformatics tools on the Galaxy platform to develop a metaproteomics workflow to characterize the metaproteomes of clinical samples. This clinical metaproteomics workflow holds potential for general clinical applications such as potential secondary infections during COVID-19 infection, microbiome changes during cystic fibrosis as well as broad research questions regarding host-microbe interactions.
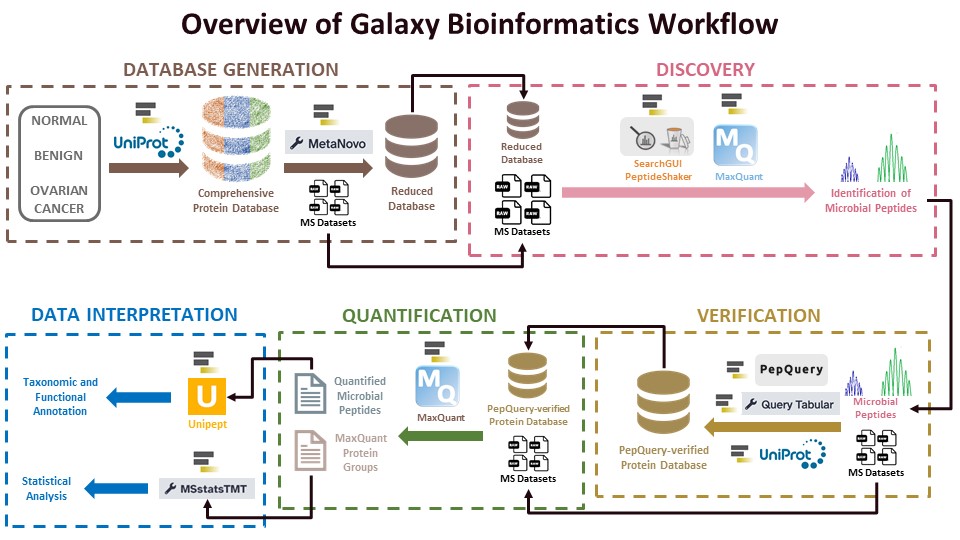
The first workflow for the clinical metaproteomics data analysis is the Database generation workflow. The Galaxy-P team has developed a workflow wherein a large database is generated by downloading protein sequences of known disease-causing microorganisms and then generating a compact database from the comprehensive database using the Metanovo tool.
Click galaxy-uploadUpload at the top of the activity panel
Select galaxy-wf-editPaste/Fetch Data
Paste the link(s) into the text field
Press Start
Close the window
As an alternative to uploading the data from a URL or your computer, the files may also have been made available from a shared data library:
Go into Libraries (left panel)
Navigate to the correct folder as indicated by your instructor.
On most Galaxies tutorial data will be provided in a folder named GTN - Material –> Topic Name -> Tutorial Name.
Select the desired files
Click on Add to Historygalaxy-dropdown near the top and select as Datasets from the dropdown menu
In the pop-up window, choose
“Select history”: the history you want to import the data to (or create a new one)
Click on Import
Rename the datasets
Check that the datatype
Click on the galaxy-pencilpencil icon for the dataset to edit its attributes
In the central panel, click galaxy-chart-select-dataDatatypes tab on the top
In the galaxy-chart-select-dataAssign Datatype, select datatypes from “New Type” dropdown
Tip: you can start typing the datatype into the field to filter the dropdown menu
Click the Save button
Optional-Add to each database a tag corresponding to the file name.
Create a dataset collection of the 4 MGF datasets.
Datasets can be tagged. This simplifies the tracking of datasets across the Galaxy interface. Tags can contain any combination of letters or numbers but cannot contain spaces.
To tag a dataset:
Click on the dataset to expand it
Click on Add Tagsgalaxy-tags
Add tag text. Tags starting with # will be automatically propagated to the outputs of tools using this dataset (see below).
Press Enter
Check that the tag appears below the dataset name
Tags beginning with # are special!
They are called Name tags. The unique feature of these tags is that they propagate: if a dataset is labelled with a name tag, all derivatives (children) of this dataset will automatically inherit this tag (see below). The figure below explains why this is so useful. Consider the following analysis (numbers in parenthesis correspond to dataset numbers in the figure below):
a set of forward and reverse reads (datasets 1 and 2) is mapped against a reference using Bowtie2 generating dataset 3;
dataset 3 is used to calculate read coverage using BedTools Genome Coverageseparately for + and - strands. This generates two datasets (4 and 5 for plus and minus, respectively);
datasets 4 and 5 are used as inputs to Macs2 broadCall datasets generating datasets 6 and 8;
datasets 6 and 8 are intersected with coordinates of genes (dataset 9) using BedTools Intersect generating datasets 10 and 11.
Now consider that this analysis is done without name tags. This is shown on the left side of the figure. It is hard to trace which datasets contain “plus” data versus “minus” data. For example, does dataset 10 contain “plus” data or “minus” data? Probably “minus” but are you sure? In the case of a small history like the one shown here, it is possible to trace this manually but as the size of a history grows it will become very challenging.
The right side of the figure shows exactly the same analysis, but using name tags. When the analysis was conducted datasets 4 and 5 were tagged with #plus and #minus, respectively. When they were used as inputs to Macs2 resulting datasets 6 and 8 automatically inherited them and so on… As a result it is straightforward to trace both branches (plus and minus) of this analysis.
Click on galaxy-workflows-activityWorkflows in the Galaxy activity bar (on the left side of the screen, or in the top menu bar of older Galaxy instances). You will see a list of all your workflows
Click on galaxy-uploadImport at the top-right of the screen
Paste the following URL into the box labelled “Archived Workflow URL”: https://training.galaxyproject.org/training-material/topics/proteomics/tutorials/clinical-mp-1-database-generation/workflows/WF1_Database_Generation_Workflow.ga
Click the Import workflow button
Below is a short video demonstrating how to import a workflow from GitHub using this procedure:
Video: Importing a workflow from URL
Run Workflowworkflow using the following parameters:
Click on Workflows on the Activity Bar on the left.
At the top of the resulting page you will have the option to switch between the My workflows, Workflows shared with me and Public workflows tabs.
Select the tab you want to see all workflows in that category
Search for your desired workflow.
Click on the workflow name: a pop-up window opens with a preview of the workflow.
To run it directly: click Run (top-right).
Recommended: click Import (left of Run) to make your own local copy under Workflows / My Workflows.
Step-by-step analysis
Download Protein Sequences using taxon names
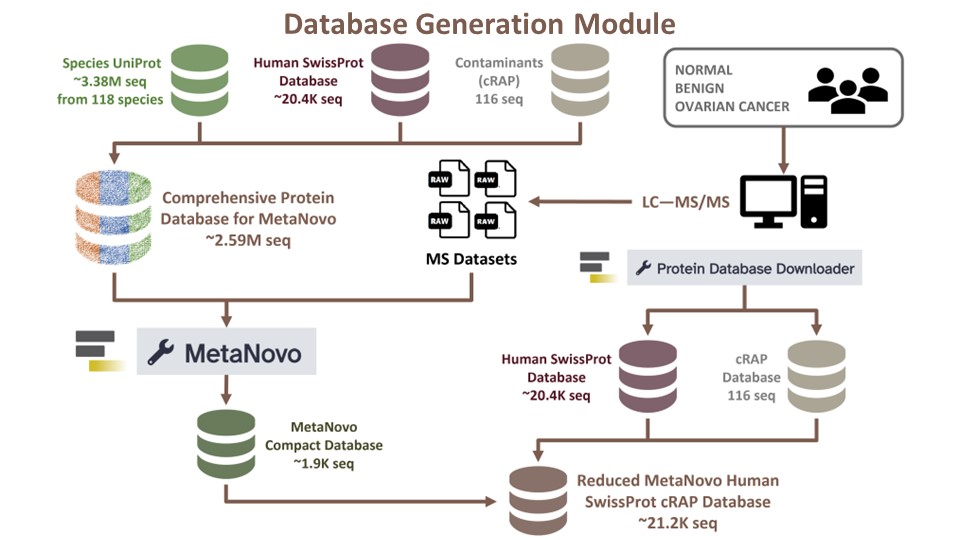
First, we want to generate a large comprehensive protein sequence database using the UniProt XML Downloader to extract sequences for species of interest. To do so, you will need a tabular file that contains a list of species.
For this tutorial, a literature survey was conducted to obtain 118 taxonomic species of organisms that are commonly associated with the female reproductive tract Afiuni-Zadeh et al. 2018. This species list was used to generate a protein sequence FASTA database was generated using the UniProt XML Downloader tool within the Galaxy framework. In this tutorial, the Species FASTA database (~3.38 million sequences) has already been provided as input. However, if you have your own list of species of interest as a tabular file (Your_Species_tabular.tabular), steps to generate a FASTA file from a tabular file are included:
Download Species Protein Sequences using UniProt XML downloader with UniProt
Hands On: UniProt XML downloader
UniProt ( Galaxy version 2.3.0) with the following parameters:
“Select”: Your_Species_tabular.tabular
param-file“Dataset (tab separated) with Taxon ID/Name column”: output (Input dataset)
“Column with Taxon ID/name”: c1
“UniProt output format”: fasta
Rename the output as Species_UniProt_FASTA.fasta
Comment: UniProt description
This tool will help download the protein fasta sequences by inputting the taxon names.
Question
Can we use a higher taxonomy clade than species for the UniProt XML downloader?
Why are we using the tools separately? Can we run it all together?
Can we select multiple files together?
How many FASTA files can be merged at once, i.e. is there a limit on the number/size of files?
Yes, the UniProt XML downloader can also be used for generating a database from Genus, Family, Order, or any other higher taxonomy clade.
The tools are run separately to reduce the load on the server and tool. If you have a limited number of taxon names, then you can run it all together.
Yes, that certainly can be done. We used one input file at a time to maintain the order of sequences in the database.
There is no limit.
Merging databases to obtain a large comprehensive database for MetaNovo
Once generated, the Species UniProt database (~3.38 million sequences) will be merged with the Human SwissProt database (reviewed only; ~20.4K sequences) and contaminant (cRAP) sequences database (116 sequences) and filtered to generate the large comprehensive database (~2.59 million sequences). The large comprehensive database will be used to generate a compact database using MetaNovo, which is much more manageable.
Hands On: Download contaminants with **Protein Database Downloader**
Protein Database Downloader ( Galaxy version 0.3.4) with the following parameters:
“Download from?”: cRAP (contaminants)
Rename as “Protein Database Contaminants (cRAP)”
Hands On: Human SwissProt (reviewed) database
Protein Database Downloader ( Galaxy version 0.3.4) with the following parameters:
“Download from?”: UniProtKB(reviewed only)
In “Taxonomy”: Homo sapiens (Human)
In “reviewed”: UniProtKB/Swiss-Prot (reviewed only)
In “Proteome Set”: Reference Proteome Set
In “Include isoform data”: False
Rename as “Protein Database Human SwissProt”.
Question
How often is the Protein Database Downloader updated?
It is updated every 3 months.
Hands On: FASTA Merge Files and Filter Unique Sequences
FASTA Merge Files and Filter Unique Sequences ( Galaxy version 1.2.0) with the following parameters:
“Run in batch mode?”: Merge individual FASTAs (output collection if input is collection)
In “Input FASTA File(s)”:
param-repeat“Insert Input FASTA File(s)”
param-file“FASTA File”: Protein Database Human SwissProt (output of Protein Database Downloadertool)
param-file“FASTA File”: Protein Database Contaminants (cRAP) (output of Protein Database Downloadertool)
param-file“FASTA File”: Species_UniProt_FASTA (output of UniProt XML downloadertool)
Rename out as “Human UniProt Microbial Proteins cRAP for MetaNovo”.
Reducing Database size
Metanovo tool generates a compact database from your comprehensive database with MetaNovo
Next, the large comprehensive database of ~2.59 million sequences can be reduced using the MetaNovo tool to generate a more manageable database that contains identified proteins.
The compact MetaNovo-generated database (~1.9K sequences) will be merged with Human SwissProt (reviewed only) and contaminants (cRAP) databases to generate the reduced database (~21.2k protein sequences) that will be used for peptide identification (see Discovery Module tutorial).
Hands On: MetaNovo
MetaNovo ( Galaxy version 1.9.4+galaxy4) with the following parameters:
param-file“FASTA File”: output (output of FASTA Merge Files and Filter Unique Sequencestool)
In “Spectrum Matching Parameters”:
“Fragment ion mass tolerance”: 0.01
“Enzyme”: Trypsin (no P rule)
“Fixed modifications as comma separated list”: Carbamidomethylation of CTMT 10-plex of KTMT 10-plex of peptide N-term
“Variable modifications as comma separated list”: Oxidation of M
“Maximal charge to search for”: 5
In “Import Filters”:
“The maximal peptide length to consider when importing identification files”: 50
Rename as “MetaNovo Compact Database”.
Question
Why are we reducing the size of the database?
Why is this running TMT10 plex modification when the data is 11-plex?
Regarding MetaNovo Spectrum Matching parameters, what are the most “important” parameters? Meaning, that if a user wants to reduce or increase the sensitivity/number of output sequences, what should they change?
Reducing the size of the database improves search speed, FDR, and sensitivity.
There is no option for 11-plex modifications in Metanovo, hence we use the TMT-10plex.
The most important parameters are the tolerance (MS1 and MS2) and any modifications introduced during the processing of the data.
Merging databases to obtain reduced MetaNovo database for peptide discovery with FASTA Merge Files and Filter Unique Sequences
Hands On: FASTA Merge Files and Filter Unique Sequences
FASTA Merge Files and Filter Unique Sequences ( Galaxy version 1.2.0) with the following parameters:
“Run in batch mode?”: Merge individual FASTAs (output collection if input is collection)
In “Input FASTA File(s)”:
param-repeat“Insert Input FASTA File(s)”
param-file“FASTA File”: Protein Database Human SwissProt (output of Protein Database Downloadertool)
param-file“FASTA File”: Protein Database Contaminants (cRAP) (output of Protein Database Downloadertool)
param-file“FASTA File”: MetaNovo Compact Database (output of MetaNovotool)
Conclusion
The first step for the Clinical Metaproteomics study is database generation. As we didn’t have a reference database or information from 16srRNA-seq data, we generated a fasta database doing a literature survey, however, if 16S rRNA data is present, the taxon identified can be used for a customized database generation. As the size of the comprehensive database is generally too large, we used the Metanovo tool to reduce the size of the database. This reduced database will be then used for clinical metaproteomics discovery workflow.
You've Finished the Tutorial
Please also consider filling out the Feedback Form as well!
Key points
Create a customized proteomics database from 16SrRNA results.
Frequently Asked Questions
Have questions about this tutorial? Have a look at the available FAQ pages and support channels
Further information, including links to documentation and original publications, regarding the tools, analysis techniques and the interpretation of results described in this tutorial can be found here.
References
Jagtap, P. D., A. Blakely, K. Murray, S. Stewart, J. Kooren et al., 2015 Metaproteomic analysis using the Galaxy framework. PROTEOMICS 15: 3553–3565. 10.1002/pmic.201500074
Afiuni-Zadeh, S., K. L. M. Boylan, P. D. Jagtap, T. J. Griffin, J. D. Rudney et al., 2018 Evaluating the potential of residual Pap test fluid as a resource for the metaproteomic analysis of the cervical-vaginal microbiome. Scientific Reports 8: 10.1038/s41598-018-29092-4
Batut, B., S. Hiltemann, A. Bagnacani, D. Baker, V. Bhardwaj et al., 2018 Community-Driven Data Analysis Training for Biology. Cell Systems 6: 752–758.e1. 10.1016/j.cels.2018.05.012
Feedback
Did you use this material as an instructor? Feel free to give us feedback on how it went.
Did you use this material as a learner or student? Click the form below to leave feedback.
Hiltemann, Saskia, Rasche, Helena et al., 2023 Galaxy Training: A Powerful Framework for Teaching! PLOS Computational Biology 10.1371/journal.pcbi.1010752
Batut et al., 2018 Community-Driven Data Analysis Training for Biology Cell Systems 10.1016/j.cels.2018.05.012
@misc{proteomics-clinical-mp-1-database-generation,
author = "Subina Mehta and Katherine Do and Dechen Bhuming",
title = "Clinical Metaproteomics 1: Database-Generation (Galaxy Training Materials)",
year = "",
month = "",
day = "",
url = "\url{https://training.galaxyproject.org/training-material/topics/proteomics/tutorials/clinical-mp-1-database-generation/tutorial.html}",
note = "[Online; accessed TODAY]"
}
@article{Hiltemann_2023,
doi = {10.1371/journal.pcbi.1010752},
url = {https://doi.org/10.1371%2Fjournal.pcbi.1010752},
year = 2023,
month = {jan},
publisher = {Public Library of Science ({PLoS})},
volume = {19},
number = {1},
pages = {e1010752},
author = {Saskia Hiltemann and Helena Rasche and Simon Gladman and Hans-Rudolf Hotz and Delphine Larivi{\`{e}}re and Daniel Blankenberg and Pratik D. Jagtap and Thomas Wollmann and Anthony Bretaudeau and Nadia Gou{\'{e}} and Timothy J. Griffin and Coline Royaux and Yvan Le Bras and Subina Mehta and Anna Syme and Frederik Coppens and Bert Droesbeke and Nicola Soranzo and Wendi Bacon and Fotis Psomopoulos and Crist{\'{o}}bal Gallardo-Alba and John Davis and Melanie Christine Föll and Matthias Fahrner and Maria A. Doyle and Beatriz Serrano-Solano and Anne Claire Fouilloux and Peter van Heusden and Wolfgang Maier and Dave Clements and Florian Heyl and Björn Grüning and B{\'{e}}r{\'{e}}nice Batut and},
editor = {Francis Ouellette},
title = {Galaxy Training: A powerful framework for teaching!},
journal = {PLoS Comput Biol}
}
Congratulations on successfully completing this tutorial!
Do you want to extend your knowledge?
Follow one of our recommended follow-up trainings:
1 stars:
Liked: Was unable to do any of the tutorial (and therefore any of the subsequent tutorials) as the links to the workflow don't work, and I can't find the workflow on the Github page :(((((
Disliked: Give a working link to the workflow
June 2024
1 stars:
Liked: https://training.galaxyproject.org/training-material/topics/proteomics/tutorials/clinical-mp-database-generation/workflows/main_workflow.ga this website (workflows) doesn't exist
Questions: