From NCBI's Sequence Read Archive (SRA) to Galaxy: SARS-CoV-2 variant analysis
| Author(s) |
|
| Editor(s) |
|
| Reviewers |
|
OverviewQuestions:
Objectives:
How can you download public sequencing data deposited in the NCBI Sequence Read Archive (SRA) into a Galaxy history for analysis?
How can you process a batch of sequencing data from several samples efficiently/in parallel with Galaxy?
Starting from raw sequenced reads of whole-genome sequenced samples of SARS-CoV-2, how can you identify mutations in the genomes of these samples?
Requirements:
Understand how Galaxy and the Sequence Read Archive interact
Be able to go from Galaxy to the Short Reach Archive, query SRA, use the SRA Run Selector to send selected metadata to Galaxy, and then import sequence data from SRA into Galaxy
Understand how collections enable processing of sequencing data in batches
Understand the analysis steps required to identify and annotate genomic mutations from sequencing data of SARS-CoV-2 samples
- Introduction to Galaxy Analyses
- slides Slides: Quality Control
- tutorial Hands-on: Quality Control
- slides Slides: Mapping
- tutorial Hands-on: Mapping
Time estimation: 1 hourSupporting Materials:Published: Jun 24, 2020Last modification: Jun 14, 2024License: Tutorial Content is licensed under Creative Commons Attribution 4.0 International License. The GTN Framework is licensed under MITpurl PURL: https://gxy.io/GTN:T00315rating Rating: 4.1 (0 recent ratings, 12 all time)version Revision: 16
The aim of this tutorial is twofold:
- It introduces general patterns of working with high-throughput sequencing data in Galaxy,
- It uses SARS-CoV-2 sequencing data analysis as a concrete example of how to identify genomic mutations from high-throughput sequencing data for a batch of viral isolates in Galaxy.
AgendaIn this tutorial, we will cover:
- Prepare analysis history and data
- Identification of mutations in the batch of SARS-Cov-2 sequencing data
- Adapter trimming with fastp
- Alignment with Map with BWA-MEM
- Remove duplicates with MarkDuplicates
- Generate alignment statistics with Samtools stats
- Realign reads with lofreq viterbi
- Add indel qualities with lofreq Insert indel qualities
- Call Variants using lofreq Call variants
- Annotate variant effects with SnpEff eff:
- Create table of variants using SnpSift Extract Fields
- Summarize data with MultiQC
- Conclusion
Prepare analysis history and data
Create a new history for this analysis
Any analysis should get its own Galaxy history. So let’s start by creating a new one:
Hands On: Prepare the Galaxy history
Create a new history for this analysis
To create a new history simply click the new-history icon at the top of the history panel:
Rename the history
- Click on galaxy-pencil (Edit) next to the history name (which by default is “Unnamed history”)
- Type the new name:
SARS-CoV-2 sequence data analysis- Click on Save
- To cancel renaming, click the galaxy-undo “Cancel” button
If you do not have the galaxy-pencil (Edit) next to the history name (which can be the case if you are using an older version of Galaxy) do the following:
- Click on Unnamed history (or the current name of the history) (Click to rename history) at the top of your history panel
- Type the new name:
SARS-CoV-2 sequence data analysis- Press Enter

Find, obtain and filter information about public sequencing data
First we need to find some good, public datasets to play with.
The Sequence Read Archive (SRA) is the primary archive of unassembled reads operated by the US National Institutes of Health (NIH). SRA is a great place to get the sequencing data that underlie publications and studies. The study we are interested in for this tutorial is one on sequencing of SARS-CoV-2 viral isolates that were obtained in the Boston area in the first months of the COVID-19 pandemic (Lemieux et al. 2021).
So lets see how we can find its raw data in the SRA, and how we can get some of that data into Galaxy to analyze it. The Acknowledgments section of the publication mentions that raw reads from the study have been deposited in the SRA under BioProject PRJNA622837 so that’s going to be our starting point.
Hands On: Find the example data in the SRA
- Go to NCBI’s SRA page by pointing your browser to https://www.ncbi.nlm.nih.gov/sra
In the search box enter
PRJNA622837[BioProject]Oh, this finds a lot of samples (more than 22,000 at the time of writing)! This is because the BioProject ID we used is that of “SARS-CoV-2 Patient Sequencing from the Broad Institute”, which has seen many more sample submissions since the published study. Let’s refine our search a bit.
Replace your previous search with
PRJNA622837[BioProject] AND RNA-Seq[Strategy]This returns only those samples from the BioProject, for which the sequencing strategy was RNA-Seq.
At the time of writing, 1908 hits are retained with this query, which is still more than the approximately 800 genomes mentioned in the publication, but we got a lot closer.
CommentThe vast majority of what has been excluded by adding
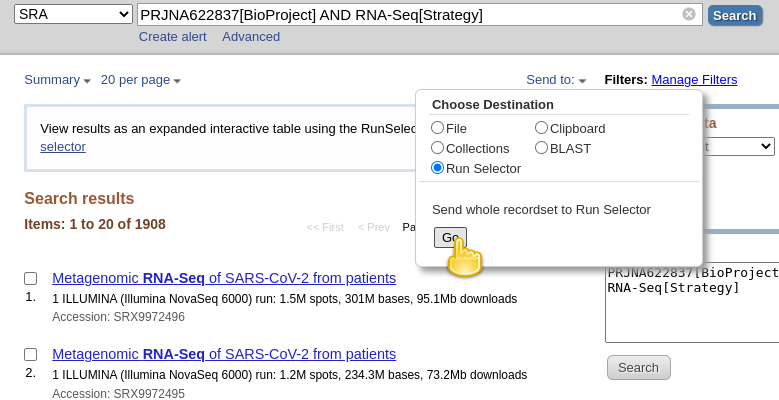
RNA-Seq[Strategy]to the query are datasets for which the “Strategy” is AMPLICON. For these datasets, the viral RNA (or more precisely, the cDNA obtained from it) got amplified with a tiled-amplicon protocol. While tiled-amplicon sequencing has emerged as a standard way of sequencing SARS-CoV-2 samples over the course of the pandemic, the corresponding protocols/primer schemes were simply not available during the early phase of the pandemic.- Instead of inspecting all 1908 hits one-by-one through the web interface, lets download the metadata for all samples in a single file
- Click on the Send to: dropdown at the top of the search results list
- Under Choose Destination, select
Run Selector- Click Go
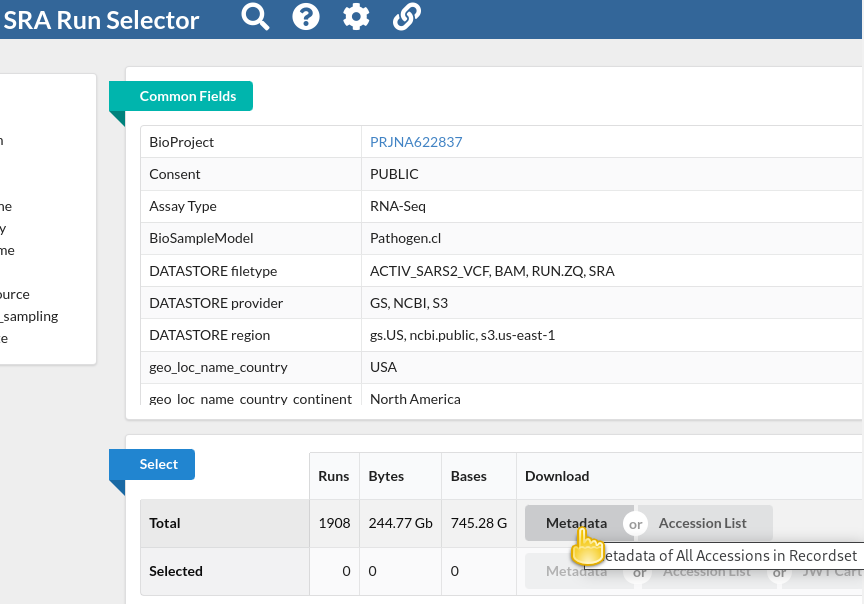
You will be taken to the SRA Run Selector page, where you can browse all of the metadata of all search hits rather conveniently.
In the Select table, click
Metadatain the Download column
This will download the metadata for all retrieved sequencing runs as a comma-separated text file.
Now that we have downloaded this file we can go to a Galaxy instance and start processing it.
CommentNote that the file we just downloaded is not sequencing data itself. Rather, it is metadata describing the data itself. We will use a few Galaxy tools to filter this list down to just a few accessions that will be used in the remainder of this tutorial.
Hands On: Upload the metadata file into Galaxy
In Galaxy, click galaxy-upload Upload Data
In the Upload dialog box, click Choose local files
- Find and select the downloaded metadata file on your computer
- Click Start
- Close the dialog by pressing Close
Once the dataset has finished uploading to your history, you can galaxy-eye inspect its contents.
You will see that this file contains the same metadata that you could preview on the SRA Run Selector page.
In Galaxy, it is rather easy to perform further filtering of the metadata records. For example, the publication mentions that the authors analyzed samples collected between 4 March and 9 May 2020.
If you inspect the metadata, you should see a column Collection_Date. That looks useful!
Hands On: Filter metadata lines by collection date range
Filter data on any column using simple expressions
- param-file “Filter”: the uploaded metadata
“With following condition”:
"2020-03-04" <= c11 <= "2020-05-09"Column 11 (c11) is the Collection_Date column in this case. If your input has this column in a different position, you would need to adjust the filter condition above accordingly.
- “Number of header lines to skip”:
1
So now we are down to sequencing runs from the same date range as studied in the publication - though we still have more runs than samples discussed in the publication. Note, however, that the publication talks about assembled genomes, and it is possible that some of the sequencing runs listed in our metadata do not contain good enough data to assemble any SARS-CoV-2 genome information, or it could be that some viral samples got sequenced in several sequencing runs to obtain enough data.
We could explore this situation in more detail, and Galaxy could also process all the sequencing runs we are currently left with, but for the sake of simplicity of the tutorial, we want to select a much smaller subset of sequencing runs now anyway. In particular, from previous experience with this data we suggest to continue with just the following two interesting datasets: SRR11954102 and SRR12733957. So let’s pull their identifiers out of the metadata file.
CommentThe following steps may seem a bit silly given that we just revealed the two identifiers above. However, think of them as placeholders for extracting run identifiers based on some other criteria.
Hands On: Extracting a subset of identifiers
Select lines that match an expression
- param-file “Select lines from”: the metadata filter by collection date; output of Filter tool
- “that”:
Matching- “the pattern”:
SRR12733957|SRR11954102Advanced Cut ( Galaxy version 1.1.0)
- param-file “File to cut”: the metadata with only two runs retained; output of Select tool
- Under “Cut by”
- “List of Fields”:
Column: 1Warning: Beware of CutsThe section below uses Cut tool. There are two cut tools in Galaxy due to historical reasons. This example uses tool with the full name Cut columns from a table. However, the same logic applies to the other tool called Advanced Cut ( Galaxy version 9.5+galaxy0). It simply has a slightly different interface.
At this point, you should have a dataset with just two lines and a single column with this content:
SRR12733957
SRR11954102
Retrieve selected sequencing data from the SRA
Now that we have our identifiers of interest extracted, we are ready to download the actual sequencing data. Since this is a very common need Galaxy offers a dedicated tool for the purpose of downloading sequencing data from the SRA identified via its run accession.
Hands On: Retrieve sequencing data from SRA via run accessions
- Faster Download and Extract Reads in FASTQ ( Galaxy version 3.0.3+galaxy0) with the following parameters:
- “select input type”:
List of SRA accession, one per line
- param-file “sra accession list”: the dataset with the two run accessions; output of Cut
Several entries should have been created in your history panel when you submitted the previous job:
- Pair-end data (fasterq-dump): Contains Paired-end datasets (if available)
- Single-end data (fasterq-dump) Contains Single-end datasets (if available)
- Other data (fasterq-dump) Contains Unpaired datasets (if available)
- fasterq-dump log Contains Information about the tool execution
The first three items are actually collections of datasets.
When the tool run is finished (all history items created by it are green / done), take a moment to explore the collections by first clicking on the collection name in the history panel. This takes you inside the collection and shows you the datasets in it. You can then navigate back to the outer level of your history.
You should find that only the Pair-end data collection contains any sequencing data, which makes sense as both samples have been paired-end sequenced on the Illumina platform.
Get the reference genome data
Since our data analysis will involve mapping the sequenced reads just downloaded to the SARS-CoV-2 reference, we also need to obtain that reference now.
Hands On: Get the reference genome data
Import the following file into your history:
https://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/009/858/895/GCF_009858895.2_ASM985889v3/GCF_009858895.2_ASM985889v3_genomic.fna.gz
- Copy the link location
Click galaxy-upload Upload at the top of the activity panel
- Select galaxy-wf-edit Paste/Fetch Data
Paste the link(s) into the text field
Press Start
- Close the window

Identification of mutations in the batch of SARS-Cov-2 sequencing data
As our actual analysis we will now perform all steps necessary to identify mutations present in the genomes of our minimal batch of two samples with respect to the SARS-CoV-2 Wuhan-Hu-1 reference sequence.
We will perform quality control and read trimming, map the reads from both samples to the reference, and perform variant calling. Finally we will annotate each sample’s variants with their predicted functional impact and report our findings in a table.
We will also summarize different quality metrics obtained throughout the analysis in one nice summary report.
Adapter trimming with fastp
Removing sequencing adapters improves alignments and variant calling. fastp tool can automatically detect widely used sequencing adapters.
Hands On: Task description
- fastp tool with the following parameters:
- “Single-end or paired reads”:
Paired Collection
- param-file “Select paired collection(s)”:
list_paired(output of Faster Download and Extract Reads in FASTQ tool)- In “Output Options”:
- “Output JSON report”:
Yes
Alignment with Map with BWA-MEM
BWA-MEM tool is a widely used sequence aligner for short-read sequencing datasets such as those we are analysing in this tutorial.
Hands On: Align sequencing reads to reference genome
- Map with BWA-MEM tool with the following parameters:
- “Will you select a reference genome from your history or use a built-in index?”:
Use a genome from history and build index
- param-file “Use the following dataset as the reference sequence”:
output(Input dataset)- “Single or Paired-end reads”:
Paired Collection
- param-file “Select a paired collection”:
output_paired_coll(output of fastp tool)- “Set read groups information?”:
Do not set- “Select analysis mode”:
1.Simple Illumina mode
Remove duplicates with MarkDuplicates
MarkDuplicates tool removes duplicate sequences originating from library preparation artifacts and sequencing artifacts. It is important to remove these artefactual sequences to avoid artificial overrepresentation of single molecule.
Hands On: Remove PCR duplicates
- MarkDuplicates tool with the following parameters:
- param-file “Select SAM/BAM dataset or dataset collection”:
bam_output(output of Map with BWA-MEM tool)- “If true do not write duplicates to the output file instead of writing them with appropriate flags set”:
Yes
Generate alignment statistics with Samtools stats
After the duplicate marking step above we can generate statistic about the alignment we have generated.
Hands On: Generate alignment statistics
- Samtools stats tool with the following parameters:
- param-file “BAM file”:
outFile(output of MarkDuplicates tool)- “Set coverage distribution”:
No- “Output”:
One single summary file- “Filter by SAM flags”:
Do not filter- “Use a reference sequence”:
No- “Filter by regions”:
No
Realign reads with lofreq viterbi
Realign reads tool corrects misalignments around insertions and deletions. This is required in order to accurately detect variants.
Hands On: Realign reads around indels
- Realign reads with lofreq tool with the following parameters:
- param-file “Reads to realign”:
outFile(output of MarkDuplicates tool)- “Choose the source for the reference genome”:
History
- param-file “Reference”:
output(Input dataset)- In “Advanced options”:
- “How to handle base qualities of 2?”:
Keep unchanged
Add indel qualities with lofreq Insert indel qualities
This step adds indel qualities into our alignment file. This is necessary in order to call variants using Call variants with lofreq tool
Hands On: Add indel qualities
- Insert indel qualities with lofreq tool with the following parameters:
- param-file “Reads”:
realigned(output of Realign reads tool)- “Indel calculation approach”:
Dindel
- “Choose the source for the reference genome”:
History
- param-file “Reference”:
output(Input dataset)
Call Variants using lofreq Call variants
We are now ready to call variants.
Hands On: Call variants
- Call variants with lofreq tool with the following parameters:
- param-file “Input reads in BAM format”:
output(output of Insert indel qualities tool)- “Choose the source for the reference genome”:
History
- param-file “Reference”:
output(Input dataset)- “Call variants across”:
Whole reference- “Types of variants to call”:
SNVs and indels- “Variant calling parameters”:
Configure settings
- In “Coverage”:
- “Minimal coverage”:
50- In “Base-calling quality”:
- “Minimum baseQ”:
30- “Minimum baseQ for alternate bases”:
30- In “Mapping quality”:
- “Minimum mapping quality”:
20- “Variant filter parameters”:
Preset filtering on QUAL score + coverage + strand bias (lofreq call default)
The output of this step is a collection of VCF files that can be visualized in a genome browser.
Annotate variant effects with SnpEff eff:
We will now annotate the variants we called in the previous step with the effect they have on the SARS-CoV-2 genome.
Hands On: Annotate variant effects
- SnpEff eff: tool with the following parameters:
- param-file “Sequence changes (SNPs, MNPs, InDels)”:
variants(output of Call variants tool)- “Output format”:
VCF (only if input is VCF)- “Create CSV report, useful for downstream analysis (-csvStats)”:
Yes- “Annotation options”: ``
- “Filter output”: ``
- “Filter out specific Effects”:
No
The output of this step is a VCF file with added variant effects.
Create table of variants using SnpSift Extract Fields
We will now select various effects from the VCF and create a tabular file that is easier to understand for humans.
Hands On: Create table of variants
- SnpSift Extract Fields tool with the following parameters:
- param-file “Variant input file in VCF format”:
snpeff_output(output of SnpEff eff: tool)- “Fields to extract”:
CHROM POS REF ALT QUAL DP AF SB DP4 EFF[*].IMPACT EFF[*].FUNCLASS EFF[*].EFFECT EFF[*].GENE EFF[*].CODON- “multiple field separator”:
,- “empty field text”:
.
Summarize data with MultiQC
We will now summarize our analysis with MultiQC, which generates a beautiful report for our data.
Hands On: Summarize data
- MultiQC tool with the following parameters:
- In “Results”:
- param-repeat “Insert Results”
- “Which tool was used generate logs?”:
fastp
- param-file “Output of fastp”:
report_json(output of fastp tool)- param-repeat “Insert Results”
- “Which tool was used generate logs?”:
Samtools
- In “Samtools output”:
- param-repeat “Insert Samtools output”
- “Type of Samtools output?”:
stats
- param-file “Samtools stats output”:
output(output of Samtools stats tool)- param-repeat “Insert Results”
- “Which tool was used generate logs?”:
Picard
- In “Picard output”:
- param-repeat “Insert Picard output”
- “Type of Picard output?”:
Markdups- param-file “Picard output”:
metrics_file(output of MarkDuplicates tool)- param-repeat “Insert Results”
- “Which tool was used generate logs?”:
SnpEff
- param-file “Output of SnpEff”:
csvFile(output of SnpEff eff: tool)
Conclusion
You have seen now how to import public sequencing data into Galaxy, and you have performed the basic steps of variant calling on a batch of RNA-Seq SARS-CoV-2 data.
Galaxy allows you to do a lot more in terms of viral sequence data analysis though. Specifically, the advanced tutorial Mutation calling, viral genome reconstruction and lineage/clade assignment from SARS-CoV-2 sequencing data is highly recommended if you want to learn how to perform a complete and more automated analysis of SARS-CoV-2 sequencing data that does not stop at lists of identified mutations but also produces consensus sequences and lineage assignments for all samples in a batch. This tutorial also shows how to deal with input data other than Illumina paired-end-sequenced RNA-Seq data.
You've Finished the Tutorial
Key points
Sequence data in the SRA can be directly imported into Galaxy
Collections enable efficient/parallel processing of sequence data from whole batches of samples
Genomic mutations in SARS-CoV-2 viral isolates can be identified through a straightforward pipeline involving sequenced reads preprocessing, mapping to the SARS-CoV-2 reference genome, postprocessing of mapping results, and variant calling
Frequently Asked Questions
Have questions about this tutorial? Have a look at the available FAQ pages and support channelsUseful literature
Further information, including links to documentation and original publications, regarding the tools, analysis techniques and the interpretation of results described in this tutorial can be found here.
References
- Lemieux, J. E., K. J. Siddle, B. M. Shaw, C. Loreth, S. F. Schaffner et al., 2021 Phylogenetic analysis of SARS-CoV-2 in Boston highlights the impact of superspreading events. Science 371: 10.1126/science.abe3261
Feedback
Did you use this material as an instructor? Feel free to give us feedback on how it went.
Did you use this material as a learner or student? Click the form below to leave feedback.

Citing this Tutorial
- Marius van den Beek, Dave Clements, Daniel Blankenberg, Anton Nekrutenko, From NCBI's Sequence Read Archive (SRA) to Galaxy: SARS-CoV-2 variant analysis (Galaxy Training Materials). https://training.galaxyproject.org/training-material/topics/variant-analysis/tutorials/sars-cov-2/tutorial.html Online; accessed TODAY
- Hiltemann, Saskia, Rasche, Helena et al., 2023 Galaxy Training: A Powerful Framework for Teaching! PLOS Computational Biology 10.1371/journal.pcbi.1010752
- Batut et al., 2018 Community-Driven Data Analysis Training for Biology Cell Systems 10.1016/j.cels.2018.05.012
Congratulations on successfully completing this tutorial!@misc{variant-analysis-sars-cov-2, author = "Marius van den Beek and Dave Clements and Daniel Blankenberg and Anton Nekrutenko", title = "From NCBI's Sequence Read Archive (SRA) to Galaxy: SARS-CoV-2 variant analysis (Galaxy Training Materials)", year = "", month = "", day = "", url = "\url{https://training.galaxyproject.org/training-material/topics/variant-analysis/tutorials/sars-cov-2/tutorial.html}", note = "[Online; accessed TODAY]" } @article{Hiltemann_2023, doi = {10.1371/journal.pcbi.1010752}, url = {https://doi.org/10.1371%2Fjournal.pcbi.1010752}, year = 2023, month = {jan}, publisher = {Public Library of Science ({PLoS})}, volume = {19}, number = {1}, pages = {e1010752}, author = {Saskia Hiltemann and Helena Rasche and Simon Gladman and Hans-Rudolf Hotz and Delphine Larivi{\`{e}}re and Daniel Blankenberg and Pratik D. Jagtap and Thomas Wollmann and Anthony Bretaudeau and Nadia Gou{\'{e}} and Timothy J. Griffin and Coline Royaux and Yvan Le Bras and Subina Mehta and Anna Syme and Frederik Coppens and Bert Droesbeke and Nicola Soranzo and Wendi Bacon and Fotis Psomopoulos and Crist{\'{o}}bal Gallardo-Alba and John Davis and Melanie Christine Föll and Matthias Fahrner and Maria A. Doyle and Beatriz Serrano-Solano and Anne Claire Fouilloux and Peter van Heusden and Wolfgang Maier and Dave Clements and Florian Heyl and Björn Grüning and B{\'{e}}r{\'{e}}nice Batut and}, editor = {Francis Ouellette}, title = {Galaxy Training: A powerful framework for teaching!}, journal = {PLoS Comput Biol} }
You can use Ephemeris's
shed-tools installcommand to install the tools used in this tutorial.shed-tools install [-g GALAXY] [-a API_KEY] -t <(curl https://training.galaxyproject.org/training-material/api/topics/variant-analysis/tutorials/sars-cov-2/tutorial.json | jq .admin_install_yaml -r)Alternatively you can copy and paste the following YAML
--- install_tool_dependencies: true install_repository_dependencies: true install_resolver_dependencies: true tools: - name: text_processing owner: bgruening revisions: d698c222f354 tool_panel_section_label: Text Manipulation tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: bwa owner: devteam revisions: 3fe632431b68 tool_panel_section_label: Mapping tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: picard owner: devteam revisions: a1f0b3f4b781 tool_panel_section_label: Picard tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: samtools_stats owner: devteam revisions: 145f6d74ff5e tool_panel_section_label: SAM/BAM tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: fastp owner: iuc revisions: dbf9c561ef29 tool_panel_section_label: FASTA/FASTQ tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: lofreq_call owner: iuc revisions: 65432c3abf6c tool_panel_section_label: Variant Calling tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: lofreq_indelqual owner: iuc revisions: 426d707dfc47 tool_panel_section_label: Variant Calling tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: lofreq_viterbi owner: iuc revisions: aa35ee7f3ab2 tool_panel_section_label: Variant Calling tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: multiqc owner: iuc revisions: bf675f34b056 tool_panel_section_label: Quality Control tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: snpeff_sars_cov_2 owner: iuc revisions: 2a3a00c1fa0a tool_panel_section_label: Variant Calling tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: snpsift owner: iuc revisions: 5fab4f81391d tool_panel_section_label: Variant Calling tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: sra_tools owner: iuc revisions: 494b2ec08162 tool_panel_section_label: Get Data tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: sra_tools owner: iuc revisions: 4df8de2d0e48 tool_panel_section_label: Get Data tool_shed_url: https://toolshed.g2.bx.psu.edu/