Galaxy-SynBioCAD portal is the first toolshed for synthetic biology, metabolic engineering, and industrial biotechnology (Hérisson et al. 2022). It provides a set of Retrosynthesis tools aimed at finding pathways to synthesize heterologous compounds in chassis organisms (RetroRules (Duigou et al. 2018), RetroPath2.0 (Delépine et al. 2018), RP2Paths, rpCompletion).
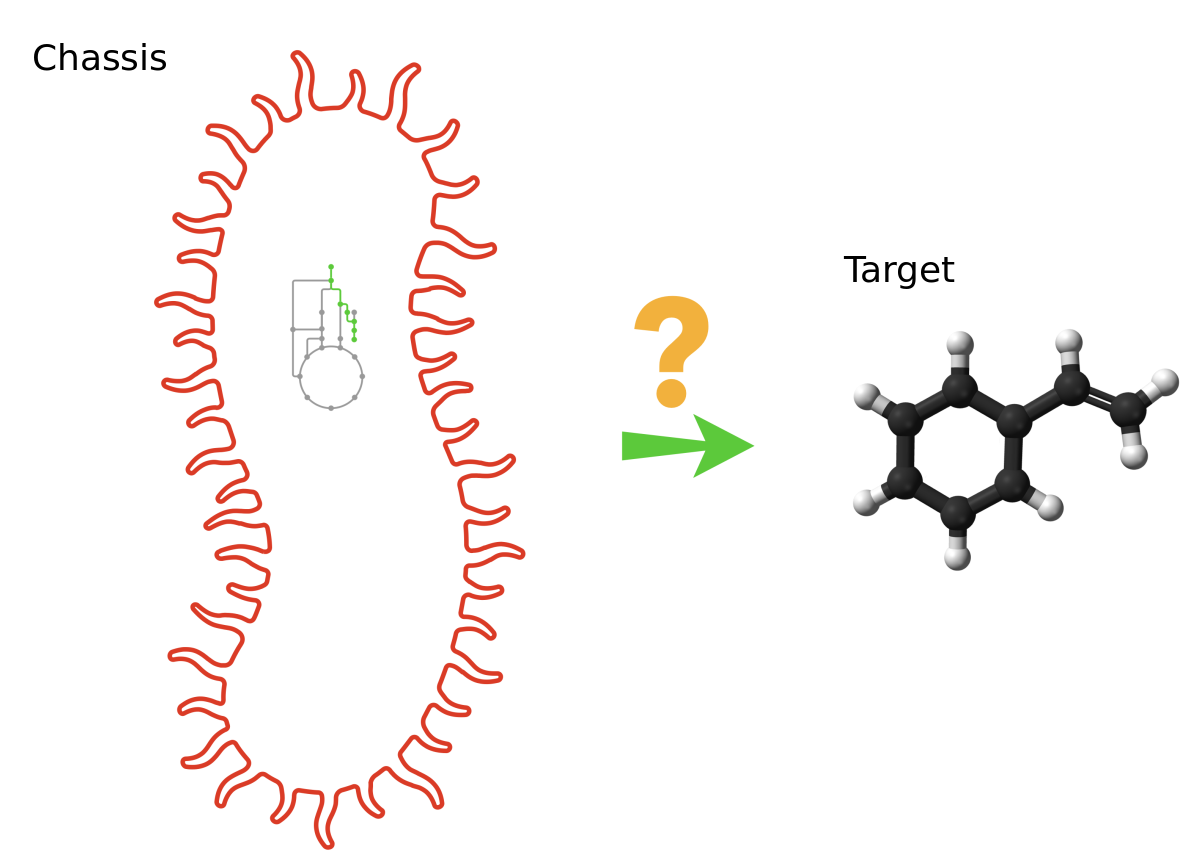
Retrosynthesis is a concept originally proposed for synthetic chemistry where chemists have to work backwards, starting from a target product to reach precursors that are endogenous to the chassis (host organism).
Typically, the target compound, also named “source compound” is the compound of interest one wishes to produce, while the precursors are usually compounds that are natively present in a chassis strain.
In this tutorial, we want to obtain the reactions producing the lycopene (source) into the iML1515 Escherichia Coli strain (chassis).
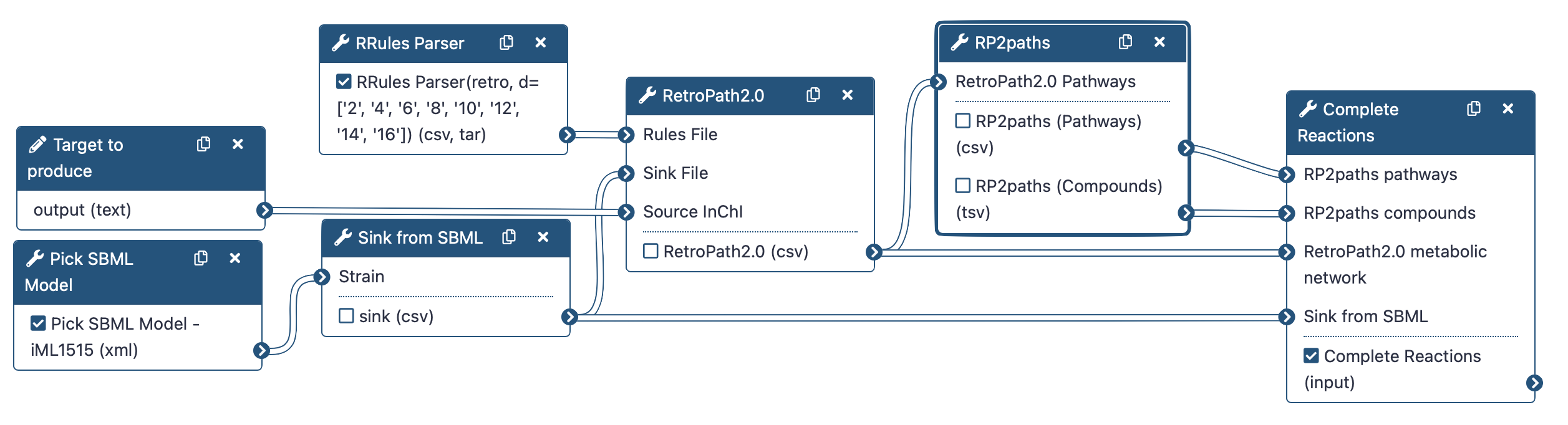
To do that, we will use the following RetroSynthetis Workflow composed of 3 key steps.
First, we aggregate the metabolites present in the chassis and download reaction rules.
Then, RetroPath2.0 generates feasible metabolic routes between a collection of chemical species contained within a GEM SBML (Systems Biology Markup Language) file of the selected organism, a target molecule that the user wishes to produce, and reaction rules extracted from RetroRules.
Lastly, the metabolic network is then deconstructed into individual pathways using RP2paths and rpCompletion takes those individual metabolic pathways to filter them (duplicated pathways are removed), splits them into sub-pathways by adding the appropriate cofactors, and finally converted them to SBML files.
Note that we will run the steps of this workflow individually so as not to neglect the understanding of the intermediate steps as well. Then, we will run the workflow automatically so that it itself retrieves the outputs from the previous step and gives them as input to the next tool.
RetroSynthesis workflow will be run with the following inputs:
The International Chemical Identifier (InChI) of the compound of interest to produce,
The structure of metabolites present in the chosen chassis (E. coli),
Reaction rules (generated by RRules Parser node that calls RetroRules).
The data used are pretty straight forward to obtain.
Firstly, we download an SBML model, then we select all sinks to use into the RetroPath2.0 software from this model.
Lastly, we request from RetroRules all possible reactions to find a chemical reaction cascade that produces the target.
Download a model
Hands On: Select a model.
Run Pick SBML Model ( Galaxy version 0.0.1) with the following parameters:
galaxy-dropdown“Strain”: Escherichia coli str. K-12 substr. MG1655 (iML1515)
Comment: What does this tool do?
The selected SBML model is downloaded from the BiGG database.
Question
What is the file format of the model?
The SBML is based on XML.
Create a sink file from the SBML model
Hands On: Generate a sink file.
Run Sink from SBML ( Galaxy version 5.12.1) with the following parameters:
param-file“Strain”: sbml_model (output of Pick SBML Modeltool)
“SBML compartment ID”: c
Comment: Choose a compartment corresponding to your model
You can specify the compartment from which the tool will extract the chemical species.
The default is c, the BiGG code for the cytoplasm.
If the user wishes to upload an SBML file from another source, then this value must be changed.
Question
What this tool does?
How many columns are in the file?
This tool creates a friendly CSV file format that can be used as sink input for RetroPath2.0.
Click on galaxy-eye, you should see 2 columns: “Name” and “InChi”
Retrieve the reaction rules
Hands On: Generate a file with all reactions.
Run RRules Parser ( Galaxy version 2.4.6) with the following parameters:
“Rule Type”: retro
“Select the diameters of the reactions rules”: 2, 4, 6, 8, 10, 12, 14 and 16
galaxy-toggle“Compress output”: no
Comment: How to choose a right diameter ?
The diameter of the sphere including the atoms around the reacting center.
The higher is the diameter, the more specific are the rules.
Question
Does a low diameter select specific rules?
How many rows are in the file?
No, a low diameter selects more unspecific rules.
More than 200 thousands!
Run the Retrosynthesis algorithm
RetroPath2.0 is an open-source tool for building retrosynthesis networks by combining reaction rules and a retrosynthesis-based algorithm to link the desired target compound to a set of available precursors.
The RetroPath2.0 tool is freely available at myExperiment.
The retrosynthesis network is outputted as a CSV file providing reactions in the reaction SMILES format and chemicals in both SMILES and InChI formats along with other information like the score for each reaction.
Launch RetroPath2.0
Hands On: Build a reaction network
Run RetroPath2.0 ( Galaxy version 2.3.0) with the following parameters:
param-file“Rules File”: out_rules (output of RRules Parsertool)
param-file“Sink File”: sink (output of Sink from SBMLtool)
Be careful, you must have InChi= in front of your InChi key.
Question
What is the file format produce by RetroPath2.0?
A csv file.
Enumerate pathways with RP2paths
The RetroPath2.0 algorithm produces a reaction network, we want to have one pathway per file in SBML format.
We need to split the network and perform some adjustments to these pathways.
Split the network
Hands On: Build a reaction network
Run RP2paths ( Galaxy version 1.5.0) with the following parameters:
param-file“RetroPath2.0 Pathways”: Reaction_Network (output of RetroPath2.0tool)
Comment: Principle
Extracts the set of pathways that lies in a metabolic space file output by the RetroPath2.0 workflow
Question
Why producing multiple pathways?
How many outputs are produced?
One network could represent several pathways, so several solutions.
Two outputs are produced: one corresponding to the metabolites, the other one corresponding to the pathways.
Refine reactions
Hands On: Refine reactions
Run Complete Reactions ( Galaxy version 5.12.2) with the following parameters:
param-file“RP2paths pathways”: master_pathways (output of RP2pathstool)
param-file“RP2paths compounds”: compounds (output of RP2pathstool)
param-file“RetroPath2.0 metabolic network”: Reaction_Network (output of RetroPath2.0tool)
param-file“Sink from SBML”: sink (output of Sink from SBMLtool)
Comment: Principle
Each reaction rule can correspond to several template reactions, the task here is to enumerate the different possible transformations according to these templates.
Because the RetroRules reaction rules consider only one substrate at a time, some compounds are by purpose omitted: the task here is to complete the predicted reactions by putting back these omitted compounds (mostly cofactors).
The node converts each predicted pathway to distinct SBML files.
Question
How many solutions are found?
In which file format are the pathways?
Do these pathways are a good solution?
We have 9 candidates.
The pathways are represented in a SBML format.
It’s a trap! We don’t know if they represent a good solution. We need to evaluate them.
Run the RetroSynthesis Workflow
In this section, you can run the RetroSynthesis Workflow more easily and fastly following these instructions:
Click on galaxy-workflows-activityWorkflows in the Galaxy activity bar (on the left side of the screen, or in the top menu bar of older Galaxy instances). You will see a list of all your workflows
Click on galaxy-uploadImport at the top-right of the screen
Paste the following URL into the box labelled “Archived Workflow URL”: https://training.galaxyproject.org/training-material/topics/synthetic-biology/tutorials/basic_assembly_analysis/workflows/Genetic_Design_BASIC_Assembly.ga
Click the Import workflow button
Below is a short video demonstrating how to import a workflow from GitHub using this procedure:
Video: Importing a workflow from URL
Click on Workflow on the top menu bar of Galaxy. You will see RetroSynthesis workflow.
Click on the workflow-run (Run workflow) button next to your workflow
Provide the workflow with the following parameters:
“Target to produce”: Provide the following Inchi source: InChI=1S/C40H56/c1-33(2)19-13-23-37(7)27-17-31-39(9)29-15-25-35(5)21-11-12-22-36(6)26-16-30-40(10)32-18-28-38(8)24-14-20-34(3)4/h11-12,15-22,25-32H,13-14,23-24H2,1-10H3/b12-11+,25-15+,26-16+,31-17+,32-18+,35-21+,36-22+,37-27+,38-28+,39-29+,40-30+
Delépine, B., T. Duigou, P. Carbonell, and J.-L. Faulon, 2018 RetroPath2.0: A retrosynthesis workflow for metabolic engineers. Metabolic Engineering 45: 158–170. 10.1016/j.ymben.2017.12.002
Duigou, T., M. du Lac, P. Carbonell, and J.-L. Faulon, 2018 RetroRules: a database of reaction rules for engineering biology. Nucleic Acids Research 47: D1229–D1235. 10.1093/nar/gky940
Hérisson, J., T. Duigou, M. du Lac, K. Bazi-Kabbaj, M. S. Azad et al., 2022 Galaxy-SynBioCAD: Automated Pipeline for Synthetic Biology Design and Engineering. 10.1101/2022.02.23.481618
Feedback
Did you use this material as an instructor? Feel free to give us feedback on how it went.
Did you use this material as a learner or student? Click the form below to leave feedback.
Hiltemann, Saskia, Rasche, Helena et al., 2023 Galaxy Training: A Powerful Framework for Teaching! PLOS Computational Biology 10.1371/journal.pcbi.1010752
Batut et al., 2018 Community-Driven Data Analysis Training for Biology Cell Systems 10.1016/j.cels.2018.05.012
@misc{synthetic-biology-retrosynthesis_analysis,
author = "Guillaume Gricourt and Thomas Duigou and Kenza Bazi-Kabbaj and Joan Hérisson and Ioana Popescu and Jean-Loup Faulon",
title = "Generating theoretical possible pathways for the production of Lycopene in E.Coli using Retrosynthesis tools (Galaxy Training Materials)",
year = "",
month = "",
day = "",
url = "\url{https://training.galaxyproject.org/training-material/topics/synthetic-biology/tutorials/retrosynthesis_analysis/tutorial.html}",
note = "[Online; accessed TODAY]"
}
@article{Hiltemann_2023,
doi = {10.1371/journal.pcbi.1010752},
url = {https://doi.org/10.1371%2Fjournal.pcbi.1010752},
year = 2023,
month = {jan},
publisher = {Public Library of Science ({PLoS})},
volume = {19},
number = {1},
pages = {e1010752},
author = {Saskia Hiltemann and Helena Rasche and Simon Gladman and Hans-Rudolf Hotz and Delphine Larivi{\`{e}}re and Daniel Blankenberg and Pratik D. Jagtap and Thomas Wollmann and Anthony Bretaudeau and Nadia Gou{\'{e}} and Timothy J. Griffin and Coline Royaux and Yvan Le Bras and Subina Mehta and Anna Syme and Frederik Coppens and Bert Droesbeke and Nicola Soranzo and Wendi Bacon and Fotis Psomopoulos and Crist{\'{o}}bal Gallardo-Alba and John Davis and Melanie Christine Föll and Matthias Fahrner and Maria A. Doyle and Beatriz Serrano-Solano and Anne Claire Fouilloux and Peter van Heusden and Wolfgang Maier and Dave Clements and Florian Heyl and Björn Grüning and B{\'{e}}r{\'{e}}nice Batut and},
editor = {Francis Ouellette},
title = {Galaxy Training: A powerful framework for teaching!},
journal = {PLoS Comput Biol}
}
Congratulations on successfully completing this tutorial!
You can use Ephemeris's shed-tools install command to install the tools used in this tutorial.
Questions: