Frequently Asked Questions
Analysis
Does MaxQuant in Galaxy support TMT, iTRAQ, etc.?
Yes, iTRAQ 4 and 8 plex; TMT 2,6,8,10,11 plex; iodoTMT6plex
For the “quantitation method” what is the default if I just leave it as “None”? Label free?
It will report raw intensity (NON-normalized) values which were not normalized like e.g. the LFQ intensities.
How many proteins can be identified and quantified in shotgun proteomics?
This is depending on the sample, the used technique(s) and the mass spectrometer. Routinely most labs obtain 4000 proteins, but with more effort 10.000 proteins could be analyzed in a single run.
If you use a mqpar file, can you include modifications that are not in the Galaxy version? For instance, propionamide (Cys alkylation by acrylamide).
No, one is limited to the modifications which are installed in MaxQuant. The mqpar only contains more parameters / options than the GUI in galaxy. Note: one must use an mqpar from the same version like MaxQuant!
Including custom modifications into MaxQuant in Galaxy?
Unfortunately the inclusion of custom modifications is not possible by the user because it requires profound changes in the underlying code. Please let us know the modification you need by creating a new issue: https://github.com/galaxyproteomics/tools-galaxyp/issues entitled MaxQuant new modification request.
MSStats: what does ‘compare groups = yes’ mean? And the comparison matrix to define the contrast between the 2 groups?
MSstats consists of three parts:
- Reading the input files and converting them into an MSstats compatible format, doing some processing of the data at the same time
- Data processing: such as protein inference (summary), log2 transformation, normalization and missing value imputation
compare groups = yes, means that the third step is performed, which is statistical analysis: Statistical modelling to find differentially abundant protein between different groups. The groups should be specified as “condition” in the annotation file and the group comparison matrix file specifies which groups to compare against each other. In the example this is quite simple because there are only 2 groups, with 3 or more groups the comparison matrix could become more complex.
What does it mean to normalize the LFQ intensities?
Median normalization typically refers to subtracting the median of all intensities within one sample from all of the intensities (e.g. Intensity of Protein A - Median of all intensities from Sample 1) , to account for measurement variations. Before normalization log2 transformation is required since many statistical tests demand that the data is actually normal distributed. (Non log intensities show very high values but have a minimum (limit of quantification) leading to a somehow right skewed distribution, after log-transformation the intensity distribution is more like a gaussian distribution. Beside the median (or median-polish) normalization there is also other e.g. the quantile normalization.
What is the advantage of breaking down protein to peptides before mass spec?
Mass spectrometry works better for peptides: LC separation and ionization is working better on peptides than on proteins and proteins generate too complex and overlaying mass spectra due to their isotopes and their mass might be shifted due to posttranslational modifications or point mutations.
When can you use (or cannot use) Match between runs in MaxQuant?
No golden rule here. For quantitative comparison of different sample groups it can be valuable to use MBR to increase the number of identified + quantified proteins in all samples and then have more proteins that occur in most of the samples to compare them.
Which isobaric labeled quantification methods does MaxQuant in Galaxy support?
The current MaxQuant version supports: iTRAQ 4 and 8 plex; TMT 2,6,8,10,11 plex; iodoTMT6plex. Includion of TMT16 plex is in preparation.
Inputs
Do I need to create collections to run MaxQuant analysis or can I use single sample inputs?
Collections are not necessary to run MaxQuant but they make the history more clean and easier to navigate. The multiple datasets options allows to select multiple files that are not part of a collection and will give the same result as with a collection as input.
Do we need a contaminant FASTA for MQ in galaxy?
Normally MaxQuant has a default contaminant fasta that we don’t have to input ourselves. MaxQuant in galaxy comes with the option to add contaminants automatically (one does not need to add contaminants to the fasta file)
Do you need to merge the databases? Because you can select multiple fasta files in MaxQuant.
For MaxQuant one does not need to merge the databases, also MaxQuant offers the function to add common contaminants to the provided fasta.
Outputs
Does MaxQuant give as output possibility the PSMs and PEPs?
Many output options, evidence & msms contain e.g. PSM or feature level info
General Questions
Can't find one of the tools for this tutorial?
To use the tools installed and available on the Galaxy server:
- At the top of the left tool panel, type in a tool name or datatype into the tool search box.
- Shorter keywords find more choices.
- Tools can also be directly browsed by category in the tool panel.
If you can’t find a tool you need for a tutorial on Galaxy, please:
- Check that you are using a compatible Galaxy server
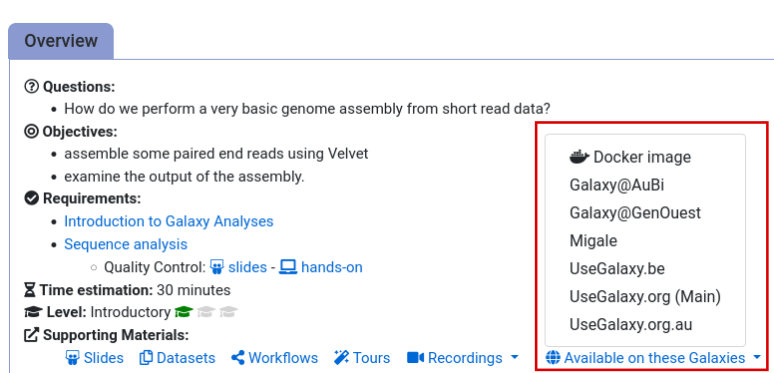
- Navigate to the overview box at the top of the tutorial
- Find the “Supporting Materials” section
- Check “Available on these Galaxies”
- If your server is not listed here, the tutorial is not supported on your Galaxy server
- You can create an account on one of the supporting Galaxies
- Use the Tutorial mode feature
- Open your Galaxy server
- Click on the curriculum icon on the top menu, this will open the GTN inside Galaxy.
- Navigate to your tutorial
- Tool names in tutorials will be blue buttons that open the correct tool for you
- Note: this does not work for all tutorials (yet)
- Still not finding the tool?
- Ask help in Gitter.
Running into an error?
When something goes wrong in Galaxy, there are a number of things you can do to find out what it was. Error messages can help you figure out whether it was a problem with one of the settings of the tool, or with the input data, or maybe there is a bug in the tool itself and the problem should be reported. Below are the steps you can follow to troubleshoot your Galaxy errors.
- Expand the red history dataset by clicking on it.
- Sometimes you can already see an error message here
View the error message by clicking on the bug icon galaxy-bug
- Check the logs. Output (stdout) and error logs (stderr) of the tool are available:
- Expand the history item
- Click on the details icon
- Scroll down to the Job Information section to view the 2 logs:
- Tool Standard Output
- Tool Standard Error
- For more information about specific tool errors, please see the Troubleshooting section
- Submit a bug report! If you are still unsure what the problem is.
- Click on the bug icon galaxy-bug
- Write down any information you think might help solve the problem
- See this FAQ on how to write good bug reports
- Click galaxy-bug Report button
- Ask for help!
- Where?
- In the GTN Matrix Channel
- In the Galaxy Matrix Channel
- Browse the Galaxy Help Forum to see if others have encountered the same problem before (or post your question).
- When asking for help, it is useful to share a link to your history