De picos a genes
| Autores/as |
|
| Traducción |
|
| Editores/as |
|
| Revisores/as |
|
Descripción GeneralPreguntas:
Objetivos:
¿Cómo utilizar Galaxy?
¿Cómo ir de zonas pico a una lista de nombres de genes
Familiarizarte con los básicos de Galaxy
Aprender cómo obtener datos de fuentes externas
Aprender cómo correr herramientas
Aprender cómo funcionan los historiales
Aprender cómo crear un workflow
Aprender a compartir tu trabajo
Duración estimada: 3 horasNivel: Introductorio IntroductoryMateriales de apoyo:
- Conjuntos de datos
- Flujos de trabajos
- galaxy-history-answer Answer Histories
- FAQs
- instances Disponible en estas instancias de Galaxy
Published: Mar 30, 2026Última modificación: Mar 30, 2026Licencia: El contenido de este tutorial tiene la licencia Creative Commons Attribution 4.0 International License. GTN Framework tiene licencia del MIT MITversion Revision: 1
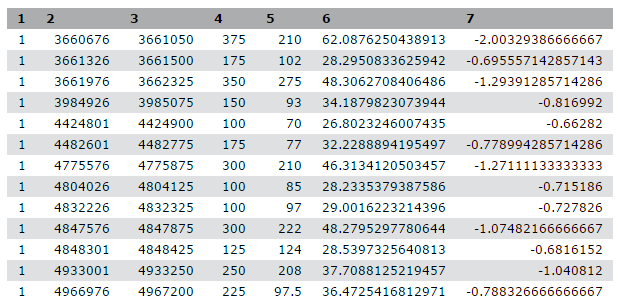
Encontramos un artículo (Li et al. 2012) titulado “The histone acetyltransferase MOF is a key regulator of the embryonic stem cell core transcriptional network“. El artículo contiene el análisis de posibles genes (llamémoslos “diana”) asociados a la proteína llamada Mof. La asociación se obtuvo mediante ChIP-seq en ratones y los datos en bruto están disponibles en GEO. Sin embargo, la lista de genes no está en el material suplementar del artículo, ni forma parte de los datos enviados a GEO. Lo más parecido que hemos podido encontrar es un archivo en [GEO](https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE37268 que contiene una lista de las regiones en las que la señal está significativamente más alta (los llamados picos)
| 1 | 3660676 | 3661050 | 375 | 210 | 62.0876250438913 | -2.00329386666667 |
| 1 | 3661326 | 3661500 | 175 | 102 | 28.2950833625942 | -0.695557142857143 |
| 1 | 3661976 | 3662325 | 350 | 275 | 48.3062708406486 | -1.29391285714286 |
| 1 | 3984926 | 3985075 | 150 | 93 | 34.1879823073944 | -0.816992 |
| 1 | 4424801 | 4424900 | 100 | 70 | 26.8023246007435 | -0.66282 |
Tabla 1 Submuestra del archivo disponible
El objetivo de este ejercicio es convertir esta lista de regiones genómicas en una lista de posibles genes diana.
Comentario: Los resultados pueden variarTus resultados pueden ser ligeramente diferentes de los presentados en este tutorial debido a las diferentes versiones de las herramientas, datos de referencia, bases de datos externas, o debido a procesos estocásticos en los algoritmos.
AgendaEn este tutorial, nos ocuparemos de:
Pretratado
Práctica: Abre Galaxy
- Navega a una instancia de Galaxy: la recomendada por tu instructor o una de la lista Instancia de Galaxy en la cabecera de esta página
Inicia sesión o regístrate (panel superior)
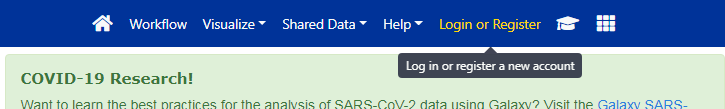
La interfaz de Galaxy consta de tres partes principales. Las herramientas disponibles se enumeran a la izquierda, su historial de análisis se registra a la derecha, y el panel central mostrará las herramientas y conjuntos de datos.
 Open image in new tab
Open image in new tabEmpecemos con una historial vacío.
Práctica: Crear historial
Asegúrate de tener un historial de análisis vacío.
Haz click sobre el icono new-history en la parte superior del panel de historiales.
Renombra tu historial para que sea fácil de reconocer
Haz clic en el título del historial (por defecto el título es
Unnamed history)
- Escribe
Galaxy Introductioncomo nombre- Pulsa Save

Carga de datos
Práctica: Carga de datos
- Descarga la lista de regiones pico (el archivo
GSE37268_mof3.out.hpeak.txt.gz) de GEO a tu ordenadorHaz clic en el botón de carga en la parte superior izquierda de la interfaz
- Pulse Choose local file y busque el archivo en su ordenador
- Seleccione
intervalcomo Type- Pulse Start
- Pulse Close
Espera a que termine la carga. Galaxy descomprimirá el archivo automáticamente.
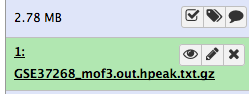
Después de esto verás tu primer elemento del historial en el panel derecho de Galaxy. Pasará por los estados gris (preparando/en cola) y amarillo (ejecutando) para convertirse en verde (éxito):
La carga directa de archivos no es la única forma de introducir datos en Galaxy
- Copia los enlaces
Abre el manejador de carga de datos de Galaxy (galaxy-upload (Upload) en la parte superior derecha del panel de herramientas)
- Selecciona ‘Pegar/Traer datos’ Paste/Fetch Data
Copia los enlaces en el campo de textos
Cambia Type (set all): de “Auto-detect” a
intervalPresiona ‘Iniciar’ Start
Close Cierra la ventana.
- Galaxy utiliza los URLs como nombres de forma predeterminada , así que los tendrás que cambiar a algunos que sean más útiles o informativos.
Hay más opciones para usuarios avanzados.
Comentario: Formato de archivo de intervaloEl formato de intervalo es un formato Galaxy para representar intervalos genómicos. Está separado por tabuladores, pero tiene el requisito añadido de que tres de las columnas deben ser:
- ID del cromosoma
- posición inicial (en base 0)
- posición final (extremo-exclusivo)
También se puede especificar una columna de cadena opcional, y se puede utilizar una fila de encabezado inicial para etiquetar las columnas, que no tienen que estar en ningún orden especial. A diferencia del formato BED (véase más adelante), también pueden aparecer columnas adicionales arbitrarias.
Puedes encontrar más información sobre los formatos que se pueden utilizar en Galaxy en la página de formatos de datos de Galaxy.
Práctica: Inspeccionar y editar atributos de un fichero
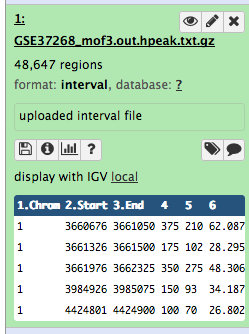
Haz clic en el archivo en el panel de la historia
A continuación se muestra alguna metainformación (por ejemplo, formato, base de datos de referencia) sobre el archivo y la cabecera del mismo, junto con el número de líneas del archivo (48.647):
Haz clic en el icono galaxy-eye (ojo) (Ver datos) en su conjunto de datos en el historial
El contenido del archivo se muestra en el panel central
Haz clic en el icono galaxy-pencil (lápiz) (Editar atributos) en su conjunto de datos en el historial
En el panel central aparece un formulario para editar los atributos del conjunto de datos
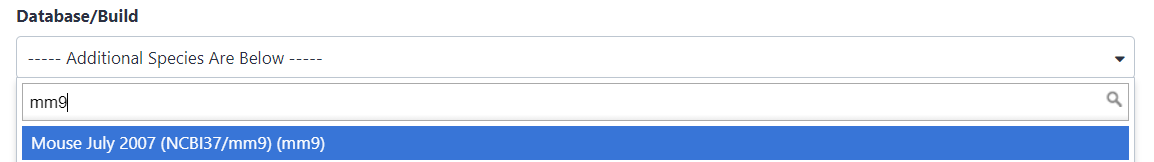
Busca
mm9en el atributo Database/Build y seleccionaMouse July 2007 (NCBI37/mm9)(el artículo nos dice que los picos son demm9)
- Haz clic en Save en la parte superior
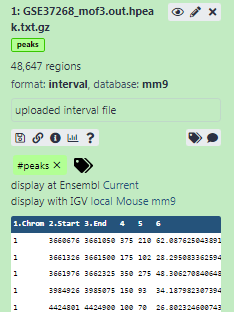
Añade una etiqueta llamada
#peaksal conjunto de datos para facilitar su seguimiento en el historialLos conjuntos de datos se pueden etiquetar. Esto simplifica el seguimiento de los conjuntos de datos a través de la interfaz de Galaxy. Las etiquetas pueden contener cualquier combinación de letras o números, pero no pueden contener espacios.
Para etiquetar un conjunto de datos:
- Haga clic en el conjunto de datos para expandirlo
- Haz click en Añadir Etiquetas galaxy-tags
- Añade tag text. Las etiquetas que empiecen por
#se propagarán automáticamente a los resultados de las herramientas que utilicen este conjunto de datos (véase más abajo).- Pulse Intro
- Compruebe que la etiqueta aparece debajo del nombre del conjunto de datos
¡Las etiquetas que empiezan por
#son especiales!Se llaman etiquetas de nombre. La característica única de estas etiquetas es que se propagan: si un conjunto de datos se etiqueta con una etiqueta de nombre, todos los derivados (hijos) de este conjunto de datos heredarán automáticamente esta etiqueta (véase más abajo). La figura siguiente explica por qué es tan útil. Considere el siguiente análisis (los números entre paréntesis corresponden a los números de los conjuntos de datos en la siguiente figura):
- un conjunto de lecturas hacia adelante y hacia atrás (conjuntos de datos 1 y 2) se mapea contra una referencia usando Bowtie2 generando el conjunto de datos 3;
- dataset 3 se utiliza para calcular la cobertura de lectura utilizando BedTools Genome Coverage por separado para las cadenas
+y-. Esto genera dos conjuntos de datos (4 y 5 para más y menos, respectivamente);- los conjuntos de datos 4 y 5 se utilizan como entrada para los conjuntos de datos Macs2 broadCall que generan los conjuntos de datos 6 y 8;
- los conjuntos de datos 6 y 8 son intersecados con las coordenadas de los genes (conjunto de datos 9) usando BedTools Intersect generando los conjuntos de datos 10 y 11.
Ahora considere que este análisis se realiza sin etiquetas de nombre. Esto se muestra en el lado izquierdo de la figura. Es difícil determinar qué conjuntos de datos contienen datos “positivos” y cuáles “negativos”. Por ejemplo, ¿el conjunto de datos 10 contiene datos “más” o datos “menos”? Probablemente “menos”, pero ¿está seguro? En el caso de un historial pequeño, como el que se muestra aquí, es posible rastrearlo manualmente, pero a medida que aumenta el tamaño del historial se convierte en todo un reto.
La parte derecha de la figura muestra exactamente el mismo análisis, pero utilizando etiquetas de nombre. Cuando se realizó el análisis, los conjuntos de datos 4 y 5 estaban etiquetados con
#plusy#minus, respectivamente. Cuando se utilizaron como entradas para Macs2, los conjuntos de datos resultantes 6 y 8 las heredaron automáticamente, y así sucesivamente… Por lo tanto, es fácil rastrear las dos ramas (positiva y negativa) de este análisis.Más información en un tutorial #nametag dedicado.
El conjunto de datos debería tener ahora el siguiente aspecto en el historial

Para encontrar los genes relacionados con estas regiones pico, también necesitamos una lista de genes en ratones, que podemos obtener de UCSC.
Práctica: Carga de datos de UCSC
Busque
UCSC Mainen la barra de búsqueda de herramientas (arriba a la izquierda)
Haz clic en
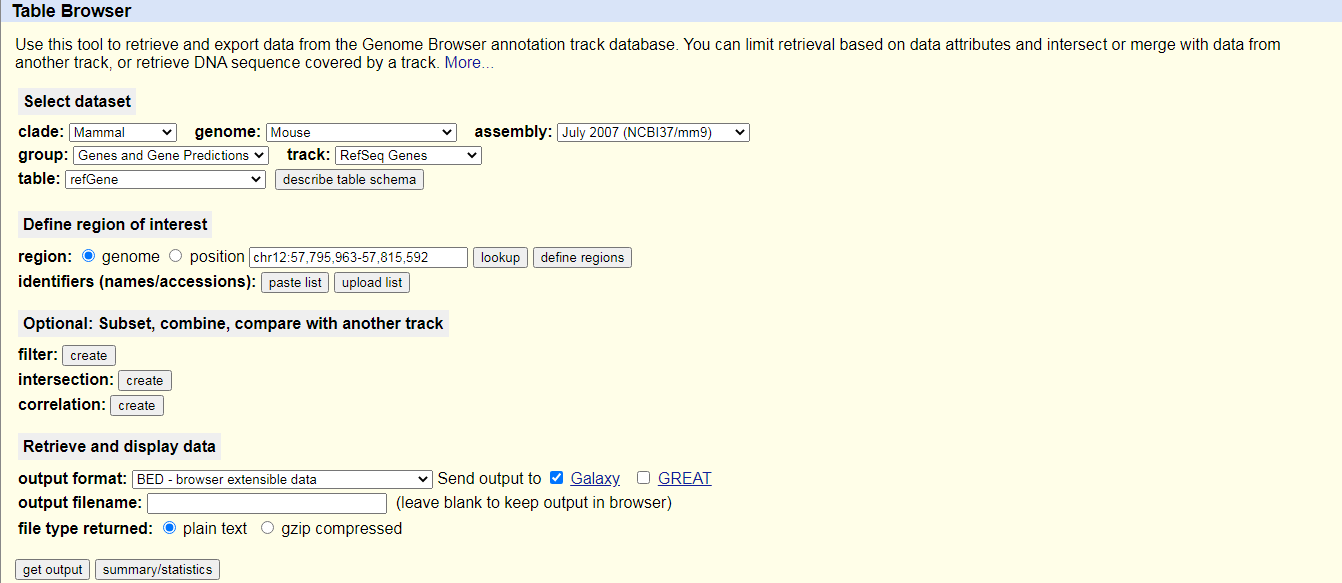
UCSC MaintoolAccederás al buscador de tablas de la UCSC, que tiene un aspecto parecido a este:
- Establece las siguientes opciones:
- “clade “:
Mammal- “genome “:
Mouse- “assembly “:
July 2007 (NCBI37/mm9)- “group “:
Genes and Gene Predictions- “track “:
RefSeq Genes- “table “:
refGene- “region “:
genome- “output format “:
BED - browser extensible data- “Send output to”:
Galaxy(sólo)Haz clic en el botón obtener salida
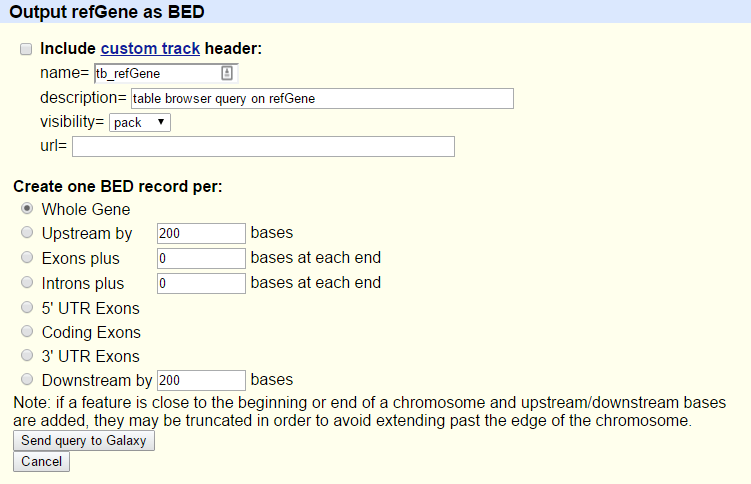
Verás la siguiente pantalla:
- Asegúrate de que “Crear un registro BED por “ está en
Whole Gene- Haz clic en el botón Enviar consulta a Galaxia
- Espera a que termine la carga
Cambia el nombre de nuestro conjunto de datos a algo más reconocible como
Genes
- Haga clic en el galaxy-pencil icono lápiz del conjunto de datos para editar sus atributos
- En el panel central, cambie el campo Name a
Genes- Haga clic en el botón Save
- Añade una etiqueta llamada
#genesal conjunto de datos para facilitar su seguimiento en el historial
Comentario: Formato de archivo BEDEl formato BED - Browser Extensible Data proporciona una forma flexible de codificar regiones de genes. Las líneas BED tienen tres campos obligatorios:
- ID del cromosoma
- posición inicial (en base 0)
- posición final (extremo-exclusivo)
Puede haber hasta nueve campos opcionales adicionales, pero el número de campos por línea debe ser coherente en cualquier conjunto de datos.
Puedes encontrar más información al respecto en UCSC, incluida una descripción de los campos opcionales.
Ahora hemos recopilado todos los datos que necesitamos para empezar nuestro análisis.
Parte 1: Enfoque ingenuo
Primero utilizaremos un enfoque “ingenuo” para intentar identificar los genes con los que están asociadas las regiones de pico. Identificaremos los genes que se solapen al menos 1bp con las regiones de pico.
Preparación del archivo
Echemos un vistazo a nuestros archivos para ver lo que realmente tenemos aquí.
Práctica: Ver contenido de archivo
Haz clic en el galaxy-eye (ojo) (Ver datos) del archivo pico para ver su contenido
Debería verse así:
Ver el contenido de las regiones de los genes de UCSC
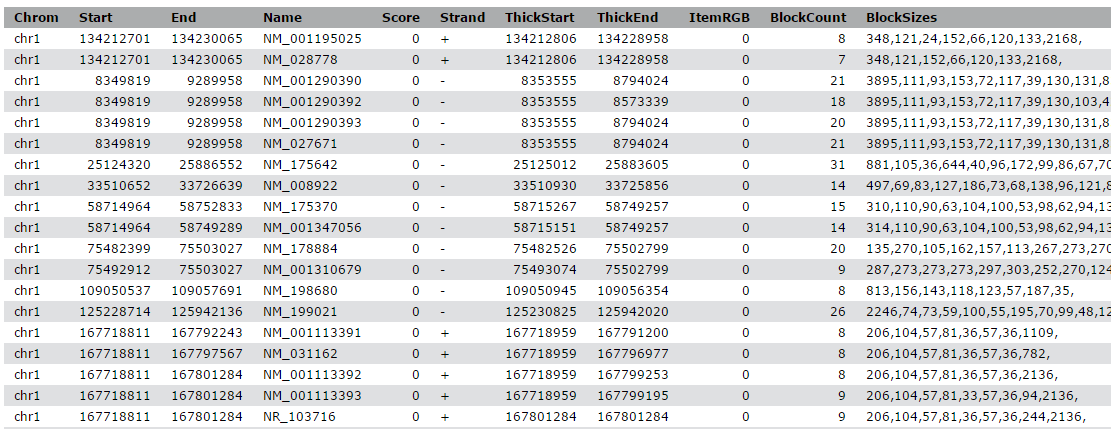
PreguntasMientras que el archivo de UCSC tiene etiquetas para las columnas, el archivo de pico no. ¿Puedes adivinar qué significan las columnas?
Este archivo de picos no tiene ningún formato estándar y, con sólo mirarlo, no podemos averiguar qué significan los números de las distintas columnas. En el artículo, los autores mencionan que utilizaron el llamador de picos HPeak.
Consultando el manual de HPeak podemos averiguar que las columnas contienen la siguiente información:
- nombre del cromosoma por número
- coordenada inicial
- coordenada final
- longitud
- ubicación dentro del pico que tiene la mayor cobertura hipotética de fragmentos de ADN (cumbre)
- no relevante
- no relevante
Para poder comparar los dos ficheros, tenemos que asegurarnos de que los nombres de los
cromosomas siguen el mismo formato. Como podemos ver, el fichero pico carece de chr
antes de cualquier número de cromosoma. Pero, ¿qué ocurre con los cromosomas 20 y 21?
¿Serán X e Y en su lugar? Comprobémoslo:
Práctica: Ver fin de archivo
- Busca Select last lines from a dataset (tail) ( Galaxy version 9.3+galaxy1) herramienta y ejecútela con la siguiente configuración:
- “Text file “: nuestro archivo pico
GSE37268_mof3.out.hpeak.txt.gz- “Operation “:
Keep last lines- “Number of lines “: Elija un valor, por ejemplo
100- Haz clic en Ejecutar herramienta
- Espera a que termine el trabajo
Inspecciona el archivo a través del galaxy-eye (ojo) icono (Ver datos)
Preguntas
- ¿Cómo se nombran los cromosomas?
- ¿Cómo se llaman los cromosomas X e Y?
- Los cromosomas se dan sólo por su número. En el archivo genético de UCSC, empezaban por
chr- Los cromosomas X e Y se llaman 20 y 21
Para convertir los nombres de los cromosomas tenemos por tanto que hacer dos cosas:
- añade
chr - cambiar 20 y 21 por X e Y
Práctica: Ajustar nombres de cromosomas
- Replace Text ( Galaxy version 1.1.3) en una columna específica con la siguiente configuración:
- “File to process “: nuestro fichero pico
GSE37268_mof3.out.hpeak.txt.gz- “in column “:
1“Find pattern “:
[0-9]+Esto buscará dígitos numéricos
“Replace with “:
chr&
&es un marcador de posición para el resultado de la búsqueda de patrónCambie el nombre de su archivo de salida
chr prefix added.- Replace Text ( Galaxy version 1.1.3) : Volvamos a ejecutar la herramienta con dos reemplazos más
- “File to process “: la salida de la última ejecución,
chr prefix added- “in column “:
1- param-repeat Reemplazar
- “Find pattern “:
chr20- “Replace with “:
chrX- param-repeat Insertar Reemplazo
- “Find pattern “:
chr21- “Replace with “:
chrY
- Ampliar la información del conjunto de datos
- Pulse el icono galaxy-refresh (Ejecute este trabajo de nuevo)
Inspecciona el último archivo a través del galaxy-eye (ojo) icono. ¿Hemos tenido éxito?
Ahora tenemos bastantes ficheros y tenemos que tener cuidado de seleccionar los correctos en cada paso.
Preguntas¿Cuántas regiones hay en nuestro archivo de salida? Puedes hacer clic en el nombre del archivo de salida para expandirlo y ver el número.
Debe ser igual al número de regiones de su primer fichero,
GSE37268_mof3.out.hpeak.txt.gz: 48,647 Si el suyo dice 100 regiones, entonces lo ha ejecutado en el archivoTaily necesita volver a ejecutar los pasos.- Cambia el nombre del archivo a algo más reconocible, por ejemplo
Peak regions
Análisis
Nuestro objetivo es comparar los 2 archivos de región (el archivo de genes y el archivo de picos de la publicación) para saber qué picos están relacionados con qué genes. Si sólo deseas saber qué picos se encuentran dentro de los genes (dentro del cuerpo del gen) puedes omitir el siguiente paso. De lo contrario, podría ser razonable incluir la región promotora de los genes en la comparación, por ejemplo, porque desea incluir factores de transcripción en experimentos ChIP-seq. No existe una definición estricta de región promotora, pero se suele utilizar 2kb upstream del TSS (inicio de la región). Usaremos la herramienta Get Flanks para obtener regiones de 2kb bases upstream del inicio del gen hasta 10kb bases downstream del inicio (12kb de longitud). Para ello, le diremos a la herramienta Get Flanks que queremos regiones upstream del inicio, con un desplazamiento de 10kb, que tengan 12kb de longitud, como se muestra en el diagrama siguiente.
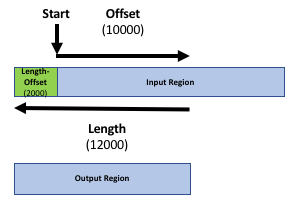
Práctica: Añadir región promotora a registros de genes
- Get Flanks ( Galaxy version 1.0.0) devuelve región/es de flanqueo para cada gen, con la siguiente configuración:
- “Select data “: archivo
Genesde UCSC- “Region “:
Around Start- “Location of the flanking region/s”:
Upstream- “Offset “:
10000- “Length of the flanking region(s):
12000Esta herramienta devuelve regiones flanqueantes para cada gen
Compara las filas del fichero BED resultante con la entrada para averiguar cómo han cambiado las posiciones de inicio y fin
Haz clic en Habilitar/Deshabilitar Scratchbook en el panel superior
- Haz clic en el galaxy-eye (ojo) de los archivos a inspeccionar
Haz clic en Show/Hide Scratchbook
- Cambia el nombre de su conjunto de datos para reflejar sus hallazgos (
Promoter regions)

La salida son regiones que empiezan a 2kb upstream del TSS e incluyen 10kb downstream. Para regiones de entrada en la cadena positiva, por ejemplo chr1 134212701
134230065, se obtiene chr1 134210701 134222701. Para regiones de la cadena negativa,
por ejemplo chr1 8349819 9289958, se obtiene chr1 9279958 9291958.
Te habrás dado cuenta de que el archivo UCSC está en formato BED y tiene asociada una
base de datos. Eso es lo que queremos también para nuestro fichero de picos. La
herramienta Intersect que utilizaremos puede convertir automáticamente los ficheros
de intervalos al formato BED, pero aquí convertiremos nuestro fichero de intervalos
explícitamente para mostrar cómo se puede conseguir esto con Galaxy.
Práctica: Cambiar formato y base de datos
- Haz clic en el icono galaxy-pencil (lápiz) en la entrada del historial de nuestro archivo de región pico
- Cambia a la pestaña Data type
- En la sección Convert to datatype en “Target datatype “ seleccione:
bed (using 'Convert Genomic Interval To Bed')- Pulse Create Dataset
- Comprueba que “Database/Build” es
mm9(la base de datos para ratones utilizada en el artículo)- De nuevo renombra el archivo a algo más reconocible, por ejemplo
Peak regions BED
Es hora de encontrar los intervalos solapados (¡por fin!). Para ello, queremos extraer los genes que se solapan/intersecan con nuestros picos.
Práctica: Encontrar solapamientos
- Intersect ( Galaxy version 1.0.0) los intervalos de dos conjuntos de datos, con la siguiente configuración:
- “Return “:
Overlapping Intervals- “of “: el archivo UCSC con regiones promotoras (
Promoter regions)- “that intersect “: nuestro archivo de región de pico de Replace (
Peak regions BED)- “for at least “:
1Comentario¡El orden de las entradas es importante! Queremos obtener una lista de genes, por lo que el conjunto de datos correspondiente con la información del gen debe ser la primera entrada (
Promoter regions).
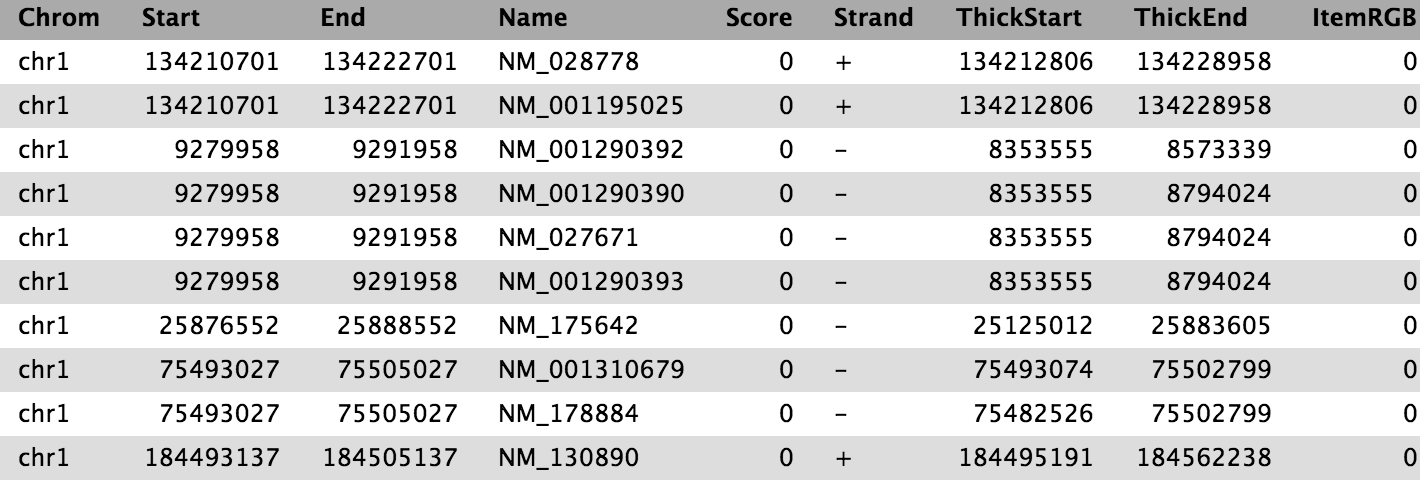
Ahora tenemos la lista de genes (columna 4) que se solapan con las regiones de pico, similar a la mostrada anteriormente.
Para tener una mejor visión de los genes que hemos obtenido, queremos ver su distribución en los diferentes cromosomas. Agruparemos la tabla por cromosomas y contaremos el número de genes con picos en cada cromosoma
Práctica: Cuenta genes en diferentes cromosomas
- Group datos por una columna y realizar la operación de agregado en otras columnas, con la siguiente configuración:
- “Select data “ al resultado de la intersección
- “Group by column “:
Column 1- Pulse Insertar operación y elija:
- “Type “:
Count- “On column “:
Column 1- “Round result to nearest integer?:
NoPreguntas¿Qué cromosoma contiene el mayor número de genes objetivo?
El resultado varía con diferentes configuraciones, por ejemplo, la anotación puede cambiar debido a actualizaciones en UCSC. Si siguió paso a paso, con la misma anotación, debería ser el cromosoma 11 con 2164 genes. Tenga en cuenta que para la reproducibilidad, debe mantener todos los datos de entrada utilizados en el análisis. Volver a ejecutar el análisis con el mismo conjunto de parámetros, almacenados Galaxy, puede conducir a un resultado diferente si las entradas cambiaron, por ejemplo, la anotación de UCSC.
Visualización
Tenemos algunos datos agregados, ¿por qué no dibujar un gráfico de barras?
Pero antes deberíamos pulir un poco más nuestros datos agrupados.
Se habrá dado cuenta de que los cromosomas del ratón no aparecen en su orden correcto en ese conjunto de datos (la herramienta Group intentó ordenarlos, pero lo hizo alfabéticamente).
Podemos arreglar esto ejecutando una herramienta dedicada para ordenar nuestros datos.
Práctica: Arreglar el orden de la tabla de recuento de genes
- Sort ( Galaxy version 1.1.1) datos en orden ascendente o descendente, con la siguiente configuración:
- “Sort query “: resultado de ejecutar la herramienta Grupo
- en param-repeat “Column selections “
- “in column “:
Column 1- “in “:
Ascending order- “Flavor “:
Natural/Version sort (-V)A veces hay varias herramientas con nombres muy parecidos. Si los parámetros del tutorial no coinciden con lo que ves en Galaxy, prueba lo siguiente:
Utiliza el Modo Tutorial curriculum en Galaxy, y haz click en el botón azul de la herramienta en el tutorial para abrir automáticamente la herramienta y versión correctas (aún no disponible para todos los tutoriales)
Las herramientas se actualizan constantemente a nuevas versiones. Tu Galaxy puede tener múltiples versiones de la misma herramienta. Por defectos, se te mostrará la última versión de cada una de ellas. Esta puede NO SER la misma herramienta utilizada en el tutorial que estás accedediendo. Además, si utilizaas una herramienta más nueva en el mismo paso… ¡puede fallar! Para asegurarte de que utilizas la misma versión de una herramienta en un tutorial concreto, utiliza el modo Tutorial mode.
- Abre tu Galaxy server
- Haz click en el icono curriculum en el menú de arriba, esto abrirá el GTN dentro de Galaxy.
- Navega hacia tu tutorial
- Los nombres de las herramientas serán botones azules que abrirán la herramienta correcta para ti
- Nota: Esto no funciona para todos los tutoriales (aún)
- Puedes clickar en cualquier parte gris fuera de la ventana del tutorial para volver a la interfície analítica de Galaxy
- Hemos tenido algunos problemas con Tutorial mode en Safari para usuarios de Mac.
- Prueba un navegador diferente si no ves el botón.
Comprueba que el nombre completo de la herramienta coincide con lo que ves en el tutorial. Compruebe que:
- Nombre completo de la herramienta:
Ordenar datos en orden ascendente o descendente- Versión de la herramienta:
1.1.1(escrita después del nombre de la herramienta)
¡Genial, estamos listos para dibujar gráficos!
Práctica: Dibujar gráfico de barras
- Haz clic en galaxy-barchart (visualizar) en la salida de la herramienta Sort
- Selecciona
Bar diagram (NVD3)- Haz clic en el « en la esquina superior derecha
- Elija un título en Provide a title, por ejemplo
Gene counts per chromosome- Cambia a la pestaña galaxy-chart-select-data Select data y juegue con la configuración
Cuando esté satisfecho, Haz clic en el galaxy-save Save en la parte superior derecha del panel principal
Esto lo almacenará en sus visualizaciones guardadas. Más tarde podrás verla, descargarla o compartirla con otros desde Data -> Visualizations en el menú superior de Galaxy.
Extrayendo un workflow
Si observamos detenidamente nuestro historial, veremos que contiene todos los pasos de nuestro análisis, desde el principio hasta el final. Al construir este historial, en realidad hemos construido un registro completo de nuestro análisis con Galaxy preservando todos los ajustes de parámetros aplicados en cada paso. ¿No sería agradable convertir este historial en un workflow que podamos ejecutar una y otra vez?
Galaxy hace esto muy simple con la opción Extract workflow. Esto significa que cada
vez que desee construir un workflow, puede realizarlo manualmente una vez, y
luego convertirlo en un workflow, de modo que la próxima vez será mucho menos
trabajo hacer el mismo análisis. También te permite compartir o publicar fácilmente tus
análisis.
Práctica: Extraer *workflow*
Limpie su historial: elimine los trabajos fallidos (en rojo) de su historial haciendo clic en el botón galaxy-delete.
Esto facilitará la creación del workflow.
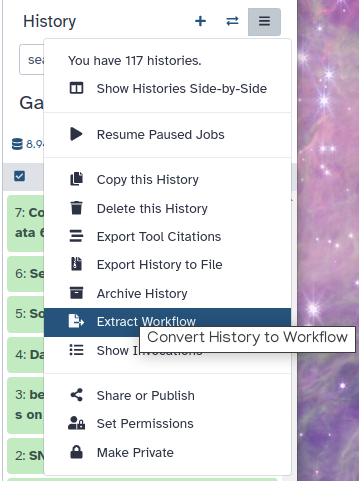
Haz clic en galaxy-gear (Opciones de historial) en la parte superior de tu panel de historial y selecciona Extract workflow.
El panel central mostrará el contenido del historial en orden inverso (el más antiguo arriba), y podrá elegir qué pasos incluir en el workflow.
Reemplace el nombre del workflow por algo más descriptivo, por ejemplo:
From peaks to genesSi hay pasos que no deberían incluirse en el workflow, puede desmarcarlos en la primera columna de casillas.
Dado que hemos realizado algunos pasos específicos para nuestro archivo de picos personalizado, es posible que queramos excluir:
- Select last tool
- todos los pasos Replace Text tool
- Convert Genomic Intervals to BED
- Get flanks tool
Haz clic en el botón Crear workflow situado en la parte superior.
Recibirá un mensaje indicando que se ha creado el workflow. Pero, ¿adónde ha ido?
Haz clic en workflow en el menú de la izquierda de Galaxy
Aquí tiene una lista de todos sus workflows
Seleccione el workflow recién generado y haz clic en Editar
Deberías ver algo similar a esto:
Comentario: El editor de *workflow*Podemos examinar el workflow en el editor de workflow de Galaxy. Aquí puede ver/cambiar la configuración de los parámetros de cada paso, añadir y eliminar herramientas, y conectar una salida de una herramienta a la entrada de otra, todo de una manera fácil y gráfica. También puedes utilizar este editor para crear workflows desde cero.
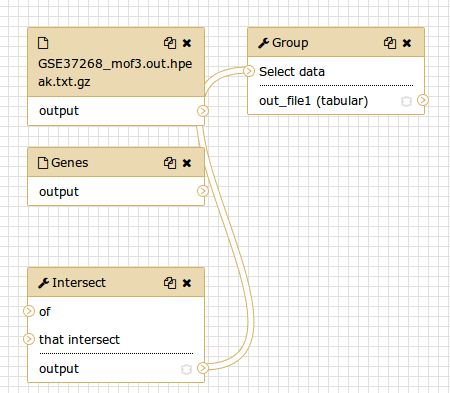
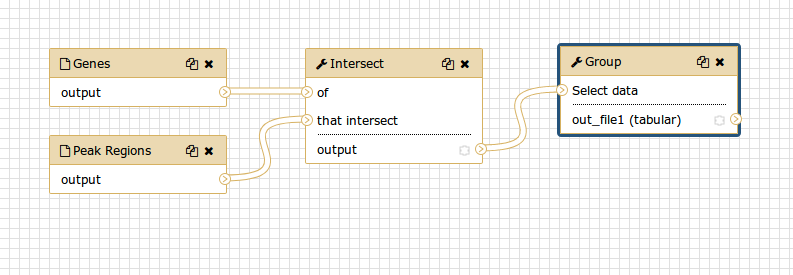
Aunque tenemos nuestros dos inputs en el workflow, no tienen la conexión a la primera herramienta (Intersect tool), porque no hemos acarreado algunos de los pasos intermedios.
- Conecta cada conjunto de datos de entrada a la herramienta Intersect tool arrastrando la flecha que apunta hacia fuera a la derecha de su casilla (que denota una salida) a una flecha a la izquierda de la casilla Intersect que apunta hacia dentro (que denota una entrada)
- Cambia el nombre de los conjuntos de datos de entrada a
Reference regionsyPeak regions- ¡Pulsa Auto Re-layout para limpiar nuestra vista
- Haz clic en el icono galaxy-save **(/training-material/topics/contributing/images/save_workflow.png){: width=”50%”}
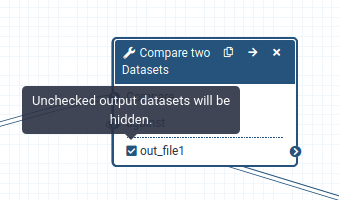
Cuando se ejecuta un workflow, el usuario suele estar interesado principalmente en el producto final y no en todos los pasos intermedios. Por defecto se mostrarán todas las salidas de un *workflow, pero podemos decirle explícitamente a Galaxy qué salida mostrar y cuál ocultar para un workflow determinado. Este comportamiento se controla mediante el pequeño asterisco que aparece junto a cada conjunto de datos de salida:
Si hace clic en este asterisco para cualquiera de los conjuntos de datos de salida, se mostrarán sólo los archivos con asterisco, y se ocultarán todas las salidas sin asterisco (Tenga en cuenta que hacer clic en todas las salidas tiene el mismo efecto que hacer clic en ninguna de las salidas, en ambos casos se mostrarán todos los conjuntos de datos).
Ahora es el momento de reutilizar nuestro workflow para un enfoque más sofisticado.
Parte 2: Enfoque más sofisticado
En la parte 1 utilizamos una definición de solapamiento de 1bp (ajuste por defecto) para identificar los genes asociados a las regiones de pico. Sin embargo, los picos podrían ser amplios, por lo que, para obtener una definición más significativa, podríamos identificar los genes que se solapan donde se concentra la mayoría de las lecturas, la cima del pico. Utilizaremos la información sobre la posición de la cumbre del pico contenida en el archivo de picos original y comprobaremos si las cumbres se solapan con los genes.
Preparación
De nuevo necesitamos nuestro archivo pico, pero nos gustaría trabajar en un historial limpio. En lugar de cargarlo dos veces, podemos copiarlo en un nuevo historial.
Práctica: Copiar elementos del historial
Crea un nuevo historial y dale un nuevo nombre como
Galaxy Introduction Part 2Haz click sobre el icono new-history en la parte superior del panel de historiales.
Haz clic en History options en la parte superior derecha de tu historial. Haz clic en Show Histories Side-by-Side
Deberías ver ahora tus dos historiales uno al lado del otro
- Arrastre y suelte el archivo de pico editado (
Peak regions, después de los pasos de reemplazo), que contiene la información de la cumbre, a su nuevo historial.- Haz clic en el nombre de la Galaxia en la barra de menú superior (arriba a la izquierda) para volver a su ventana de análisis
Crear archivo cumbre pico
Necesitamos generar un nuevo archivo BED a partir del archivo de picos original que
contenga las posiciones de las cumbres de los picos. El inicio de la cima es el inicio
del pico (columna 2) más la ubicación dentro del pico que tiene la mayor cobertura
hipotética de fragmentos de ADN (columna 5, redondeada al siguiente entero más pequeño
porque algunas cimas de picos caen entre a bases). Como final de la región del pico,
definiremos simplemente start + 1.
Práctica: Crear archivo cumbre
- Compute on rows ( Galaxy version 2.0) con los siguientes parámetros:
- “Archivo de entrada “: nuestro archivo de pico
Peak regions(el archivo de formato de intervalo)- *“¿La entrada tiene una línea de encabezado con nombres de columnas?”:
No- En “Expressions “:
- param-repeat “Expressions “
- “Add expression “:
c2 + int(c5)- “Mode of the operation “: Append
- param-repeat “Expressions “
- “Add expression “:
c8 + 1- “Mode of the operation “: Append
Esto creará una 8ª y una 9ª columna en nuestra tabla, que utilizaremos en el siguiente paso:
- Renombra la salida a
Peak summit regions
Ahora recortamos sólo el cromosoma más el inicio y el final de la cima:
Práctica: Cortar columnas
- Cut columnas de una tabla con la siguiente configuración:
- “Cut columns “:
c1,c8,c9- “Delimited by tab “:
Tab- “From “:
Peak summit regionsLa salida de Cut estará en formato
tabular.
Cambia el formato a
interval(usa el galaxy-pencil) ya que es lo que espera la herramienta Intersect.
- Selecciona sobre el galaxy-pencil icono del lápiz para editar los atributos del conjunto de datos
- Selecciona en la pestaña galaxy-chart-select-data Datatypes en la parte superior del panel central
- Selecciona
interval- Da clic en el botón Change datatype
El resultado debería ser como el siguiente:
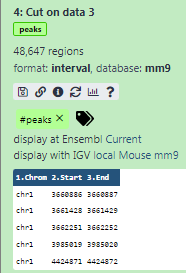
Obtener nombres de genes
Los genes RefSeq que descargamos de UCSC sólo contenían los identificadores RefSeq, pero no los nombres de los genes. Para obtener al final una lista de nombres de genes, utilizamos otro archivo BED de las Bibliotecas de Datos.
ComentarioHay varias formas de obtener los nombres de los genes, si necesita hacerlo usted mismo. Una forma es recuperar un mapeo a través de Biomart y luego unir los dos archivos (Join two Datasets side by side on a specified field tool). Otra forma es obtener la tabla RefSeq completa de UCSC y convertirla manualmente al formato BED.
Práctica: Carga de datos
Import
mm9.RefSeq_genes_from_UCSC.bedde Zenodo o de la biblioteca de datos:https://zenodo.org/record/1025586/files/mm9.RefSeq_genes_from_UCSC.bed
- Copia los enlaces
Abre el manejador de carga de datos de Galaxy (galaxy-upload (Upload) en la parte superior derecha del panel de herramientas)
- Selecciona ‘Pegar/Traer datos’ Paste/Fetch Data
Copia los enlaces en el campo de textos
Cambia Genome a
mm9Presiona ‘Iniciar’ Start
Close Cierra la ventana.
- Galaxy utiliza los URLs como nombres de forma predeterminada , así que los tendrás que cambiar a algunos que sean más útiles o informativos.
Como alternativa a cargar los datos desde una URL o desde su ordenador, los archivos también pueden estar disponibles desde una biblioteca de datos compartidos:
- Entra en Libraries (panel izquierdo)
- Navega a : Haz clic en “GTN - Material”, “Introduction to Galaxy Analyses”, “From peaks to genes”, y luego “DOI: 10.5281/zenodo.1025586” o la carpeta correcta indicada por su instructor.
- Seleccione los archivos deseados
- Haz clic en Add to History galaxy-dropdown cerca de la parte superior y selecciona as Datasets en el menú desplegable
- En la ventana emergente, elige
- “Seleccionar historial “: el historial al que desea importar los datos (o crear uno nuevo)
- Haga clic en Import
Por defecto, Galaxy toma el enlace como nombre, así que renómbralos.
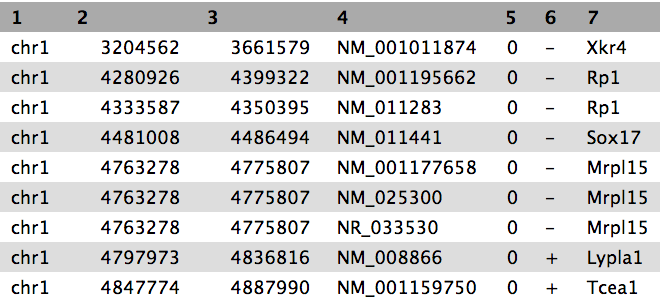
Inspeccione el contenido del archivo para comprobar si contiene nombres de genes. Debe ser similar al siguiente:
- Renómbralo
mm9.RefSeq_genes- Aplica la etiqueta
#genes
Repetir workflow
Es hora de reutilizar el workflow que creamos antes.
Práctica: Ejecutar un workflow
- Abre el menú de workflow (barra de menú izquierda)
- Busque el workflow que realizó en la sección anterior y seleccione la opción Execute
- Elige como entradas nuestro fichero BED
mm9.RefSeq_genes(#genes) y el resultado de la herramienta Cut (#peaks)Haz clic en Execute workflow
Las salidas deberían aparecer en el historial, pero puede que tarden un poco en terminar.
Usamos nuestro workflow para volver a ejecutar nuestro análisis con las cimas de los picos. La herramienta Group volvió a producir una lista con el número de genes encontrados en cada cromosoma. ¿Pero no sería más interesante conocer el número de picos en cada gen único? Volvamos a ejecutar el workflow con una configuración diferente
Práctica: Ejecutar un workflow con la configuración cambiada
- Abre el menú de workflow (barra de menú izquierda)
- Busque el workflow que realizó en la sección anterior y seleccione la opción Ejecutar
- Elige como entradas nuestro fichero BED
mm9.RefSeq_genes(#genes) y el resultado de la herramienta Cut (#peaks)- Haz clic en el título de la herramienta tool Group para ampliar las opciones.
- Cambie los siguientes ajustes haciendo clic en el galaxy-pencil (lápiz) de la izquierda:
- “Group by column”:
7- En “Operation “:
- “In column “:
7- Haz clic en Execute workflow
¡Enhorabuena! Debería tener un archivo con todos los nombres de genes únicos y un recuento de cuántos picos contenían.
PreguntasLa lista de genes únicos no está ordenada. Intente ordenarla usted mismo
Puede utilizar la herramienta “Sort data in ascending or descending order” en la columna 2 y “fast numeric sort”.
Comparte tu trabajo
Una de las características más importantes de Galaxy llega al final de un análisis. Cuando haya publicado resultados sorprendentes, es importante que otros investigadores puedan reproducir su experimento in-silico. Galaxy permite a los usuarios compartir fácilmente sus workflows e historiales con otros.
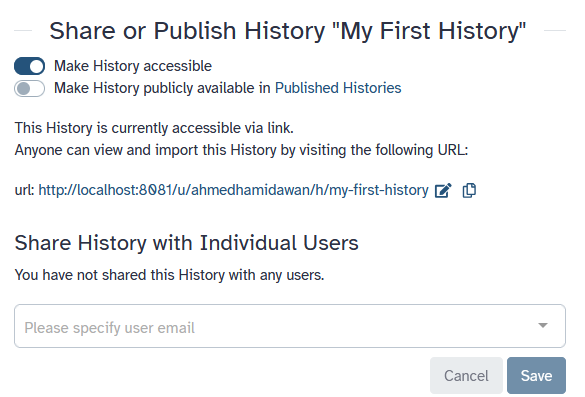
Para compartir un historial, haz clic en las galaxy-history-options opciones
de historial y selecciona Share or Publish. En esta página puedes hacer 3 cosas:
-
Hacer accesible vía Enlace
Esto genera un enlace que puedes dar a otros. Cualquiera que tenga este enlace podrá ver tu historial.
-
Hacer pública la Historia en Historias Publicadas
Esto no sólo creará un enlace, sino que también publicará tu historial. Esto significa que su historial aparecerá bajo
Data → Histories → Published Historiesen el menú superior. -
Compartir con usuarios individuales
Esto compartirá el historial sólo con usuarios específicos en la instancia Galaxy.
Práctica: Compartir historia y *workflow*
- Comparte una de tus historias con tu vecino
- ¡A ver si puedes hacer lo mismo con tu workflow!
Encuentra el historial y/o workflow compartido por tu vecino
Los historiales compartidos con usuarios específicos pueden ser accedidos por esos usuarios bajo
Data → Histories → Histories shared with me.
Conclusión
trophy Acabas de realizar su primer análisis en Galaxy. También has creado un workflow a partir de su análisis para poder repetir fácilmente el mismo análisis en otros conjuntos de datos. Además, ha compartido sus resultados y métodos con otras personas.
You've finished the tutorial
Puntos clave
Galaxy proporciona una interfaz gráfica fácil de usar para herramientas complejas de línea de comandos
Galaxy guarda un registro completo de tu análisis en un historial
Los workflows te permiten repetir tu análisis con distintos datos
Galaxy puede conectarse a fuentes externas para importar datos y visualizaciones
Galaxy ofrece formas de compartir tus resultados y métodos con otros
Preguntas frecuentes
Have questions about this tutorial? Have a look at the available FAQ pages and support channelsReferencias
- Li, X., L. Li, R. Pandey, J. S. Byun, K. Gardner et al., 2012 The Histone Acetyltransferase MOF Is a Key Regulator of the Embryonic Stem Cell Core Transcriptional Network. Cell Stem Cell 11: 163–178. 10.1016/j.stem.2012.04.023
Retroalimentación
¿Utilizaste este material como instructor? Cuéntanos tu experiencia.
¿Has usado este material como aprendiz o estudiante? Haz click en el formulario a continuación para dejarnos tu opinión

Cómo citar este tutorial
- Anne Pajon, Clemens Blank, Bérénice Batut, Björn Grüning, Nicola Soranzo, Dilmurat Yusuf, Sarah Peter, Helena Rasche, De picos a genes (Galaxy Training Materials). https://training.galaxyproject.org/training-material/topics/introduction/tutorials/galaxy-intro-peaks2genes/tutorial_ES.html Online; accessed TODAY
- Hiltemann, Saskia, Rasche, Helena et al., 2023 Galaxy Training: A Powerful Framework for Teaching! PLOS Computational Biology 10.1371/journal.pcbi.1010752
- Batut et al., 2018 Community-Driven Data Analysis Training for Biology Cell Systems 10.1016/j.cels.2018.05.012
@misc{introduction-galaxy-intro-peaks2genes, author = "Anne Pajon and Clemens Blank and Bérénice Batut and Björn Grüning and Nicola Soranzo and Dilmurat Yusuf and Sarah Peter and Helena Rasche", title = "De picos a genes (Galaxy Training Materials)", year = "", month = "", day = "", url = "\url{https://training.galaxyproject.org/training-material/topics/introduction/tutorials/galaxy-intro-peaks2genes/tutorial_ES.html}", note = "[Online; accessed TODAY]" } @article{Hiltemann_2023, doi = {10.1371/journal.pcbi.1010752}, url = {https://doi.org/10.1371%2Fjournal.pcbi.1010752}, year = 2023, month = {jan}, publisher = {Public Library of Science ({PLoS})}, volume = {19}, number = {1}, pages = {e1010752}, author = {Saskia Hiltemann and Helena Rasche and Simon Gladman and Hans-Rudolf Hotz and Delphine Larivi{\`{e}}re and Daniel Blankenberg and Pratik D. Jagtap and Thomas Wollmann and Anthony Bretaudeau and Nadia Gou{\'{e}} and Timothy J. Griffin and Coline Royaux and Yvan Le Bras and Subina Mehta and Anna Syme and Frederik Coppens and Bert Droesbeke and Nicola Soranzo and Wendi Bacon and Fotis Psomopoulos and Crist{\'{o}}bal Gallardo-Alba and John Davis and Melanie Christine Föll and Matthias Fahrner and Maria A. Doyle and Beatriz Serrano-Solano and Anne Claire Fouilloux and Peter van Heusden and Wolfgang Maier and Dave Clements and Florian Heyl and Björn Grüning and B{\'{e}}r{\'{e}}nice Batut and}, editor = {Francis Ouellette}, title = {Galaxy Training: A powerful framework for teaching!}, journal = {PLoS Comput Biol} }
Referencias
These individuals or organisations provided funding support for the development of this resource
¡Felicitaciones! ¡Completaste con éxito este tutorial!

You can use Ephemeris's
shed-tools installcommand to install the tools used in this tutorial.shed-tools install [-g GALAXY] [-a API_KEY] -t <(curl https://training.galaxyproject.org/training-material/api/topics/introduction/tutorials/galaxy-intro-peaks2genes/tutorial.json | jq .admin_install_yaml -r)Alternatively you can copy and paste the following YAML
--- install_tool_dependencies: true install_repository_dependencies: true install_resolver_dependencies: true tools: - name: text_processing owner: bgruening revisions: d698c222f354 tool_panel_section_label: Text Manipulation tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: text_processing owner: bgruening revisions: c41d78ae5fee tool_panel_section_label: Text Manipulation tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: text_processing owner: bgruening revisions: c41d78ae5fee tool_panel_section_label: Text Manipulation tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: text_processing owner: bgruening revisions: d698c222f354 tool_panel_section_label: Text Manipulation tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: text_processing owner: bgruening revisions: c41d78ae5fee tool_panel_section_label: Text Manipulation tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: column_maker owner: devteam revisions: aff5135563c6 tool_panel_section_label: Text Manipulation tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: get_flanks owner: devteam revisions: 077f404ae1bb tool_panel_section_label: Text Manipulation tool_shed_url: https://toolshed.g2.bx.psu.edu/ - name: intersect owner: devteam revisions: 69c10b56f46d tool_panel_section_label: Text Manipulation tool_shed_url: https://toolshed.g2.bx.psu.edu/