Frequently Asked Questions
Tutorial Questions
Are these data free to use and download?
Question: Are these data free to use and download?Yes, the metadata, aligned reads, and other SARS-CoV-2 data that is mentioned in this training are free to download and have no associated egress charges.
Do you have resources to help me get started working in the cloud?
Question: Do you have resources to help me get started working in the cloud?Yes, we have a number of documents and videos to help you start working with SRA data in the cloud:
When will aligned read objects be available for other data types?
Question: When will aligned read objects be available for other data types?We hope to have these constructed for long read SARS-CoV-2 data in the near future. If there is strong community interest we may expand this offering to other organisms or data types such as metagenome submissions. If you would like this format for other datasets, write to the SRA helpdesk (sra@ncbi.nlm.nih) and let us know!
Where can I find example queries for use in the cloud and elsewhere?
Question: Where can I find example queries for use in the cloud and elsewhere?We have examples on our website for Athena (link) and BigQuery (link) which can be easily adapted to other environments.
Where can I find the full listing and description of the columns in each metadata table?
Question: Where can I find the full listing and description of the columns in each metadata table?Table definitions are available here:
Why don't the aligned read files have quality scores?
Question: Why don't the aligned read files have quality scores?Quality scores take up the majority of space in our compressed sequence files, so removing them makes the files much smaller (~80% or more). In addition, many uses don’t require per-base quality scores to successfully complete their work (some pipelines even require fastq format but don’t actually use the quality scores), so these files represent a faster route to completing many analyses. The full quality scores are still available in the original SRA Runs for anyone that requires them, using the SRA Tools available in Galaxy.
General Questions
Can't find one of the tools for this tutorial?
To use the tools installed and available on the Galaxy server:
- At the top of the left tool panel, type in a tool name or datatype into the tool search box.
- Shorter keywords find more choices.
- Tools can also be directly browsed by category in the tool panel.
If you can’t find a tool you need for a tutorial on Galaxy, please:
- Check that you are using a compatible Galaxy server
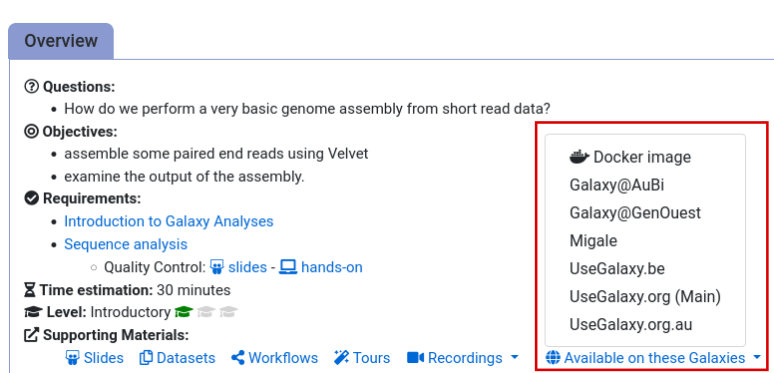
- Navigate to the overview box at the top of the tutorial
- Find the “Supporting Materials” section
- Check “Available on these Galaxies”
- If your server is not listed here, the tutorial is not supported on your Galaxy server
- You can create an account on one of the supporting Galaxies
- Use the Tutorial mode feature
- Open your Galaxy server
- Click on the curriculum icon on the top menu, this will open the GTN inside Galaxy.
- Navigate to your tutorial
- Tool names in tutorials will be blue buttons that open the correct tool for you
- Note: this does not work for all tutorials (yet)
- Still not finding the tool?
- Ask help in Gitter.
Running into an error?
When something goes wrong in Galaxy, there are a number of things you can do to find out what it was. Error messages can help you figure out whether it was a problem with one of the settings of the tool, or with the input data, or maybe there is a bug in the tool itself and the problem should be reported. Below are the steps you can follow to troubleshoot your Galaxy errors.
- Expand the red history dataset by clicking on it.
- Sometimes you can already see an error message here
View the error message by clicking on the bug icon galaxy-bug
- Check the logs. Output (stdout) and error logs (stderr) of the tool are available:
- Expand the history item
- Click on the details icon
- Scroll down to the Job Information section to view the 2 logs:
- Tool Standard Output
- Tool Standard Error
- For more information about specific tool errors, please see the Troubleshooting section
- Submit a bug report! If you are still unsure what the problem is.
- Click on the bug icon galaxy-bug
- Write down any information you think might help solve the problem
- See this FAQ on how to write good bug reports
- Click galaxy-bug Report button
- Ask for help!
- Where?
- In the GTN Matrix Channel
- In the Galaxy Matrix Channel
- Browse the Galaxy Help Forum to see if others have encountered the same problem before (or post your question).
- When asking for help, it is useful to share a link to your history