Frequently Asked Questions
Tutorial Questions
Automatically trim adapters (without providing custom sequences)
There are many tools for this: Trimmomatic, Trim Galore, and a few others (search: “Trim”). In some of these there are options to automatically trim adaptors, but they are not so specific to the sequence you are working on necessarily.
Changing the heatmap colours
You can change the heatmap color, by expanding the Show advanced options section. There are many options here, including setting the colors.
In bowtie 2 parameters, in place of 1000 for other experiments, should we mention the median fragment length observed in our library?
Not the median fragment length but the maximum fragment length you expect. However, you will see that in illumina sequencers, the longer the fragments are the less efficiently they are sequenced so long fragment length pairs are not very numerous.
Is it possible to subsample some samples if you have more reads?
Yes, we would recommend to process all reads and just before the peak calling. You can use tool Samtools view to sample the BAM file.
UCSC import: what should my file look like?
~2020 lines, with the following header line:
bin name chrom strand txStart txEnd cdsStart cdsEnd exonCount exonStarts exonEnds score name2 cdsStartStat cdsEndStat exonFramesWhere:
txStart: Transcript start sitecdsStart: CodingSequence start siteNote: UCSC is updated frequently, you might get a slightly different number of lines. If you only get one row in this file, make sure you requested the entire chr22, not just one position.
Datatypes
Compressed FASTQ files, (`*.gz`)
- Files ending in
.gzare compressed (zipped) files.
- The
fastq.gzformat is a compressed version of afastqdataset.- The
fastqsanger.gzformat is a compressed version of thefastqsangerdatatype, etc.- Compression saves space (and therefore your quota).
- Tools can accept the compressed versions of input files
- Make sure the datatype (compressed or uncompressed) is correct for your files, or it may cause tool errors.
FASTQ files: `fastq` vs `fastqsanger` vs ..
FASTQ files come in various flavours. They differ in the encoding scheme they use. See our QC tutorial for a more detailed explanation of encoding schemes.
Nowadays, the most commonly used encoding scheme is sanger. In Galaxy, this is the
fastqsangerdatatype. If you are using older datasets, make sure to verify the FASTQ encoding scheme used in your data.Be Careful: choosing the wrong encoding scheme can lead to incorrect results!
Tip: There are 2 Galaxy datatypes that have similar names, but are not the same, please make sure you
fastqsangerandfastqcssanger(not the additionalcs).Tip: When in doubt, choose
fastqsanger
Visualisation
Using IGV with Galaxy
You can send data from your Galaxy history to IGV for viewing as follows:
- Install IGV on your computer (IGV download page)
- Start IGV
- In recent versions of IGV, you will have to enable the port:
- In IGV, go to
View > Preferences > Advanced- Check the box
Enable Port- In Galaxy, expand the dataset you would like to view in IGV
- Make sure you have set a reference genome/database correctly (dbkey) (instructions)
- Under
display in IGV, click onlocal
General Questions
Can't find one of the tools for this tutorial?
To use the tools installed and available on the Galaxy server:
- At the top of the left tool panel, type in a tool name or datatype into the tool search box.
- Shorter keywords find more choices.
- Tools can also be directly browsed by category in the tool panel.
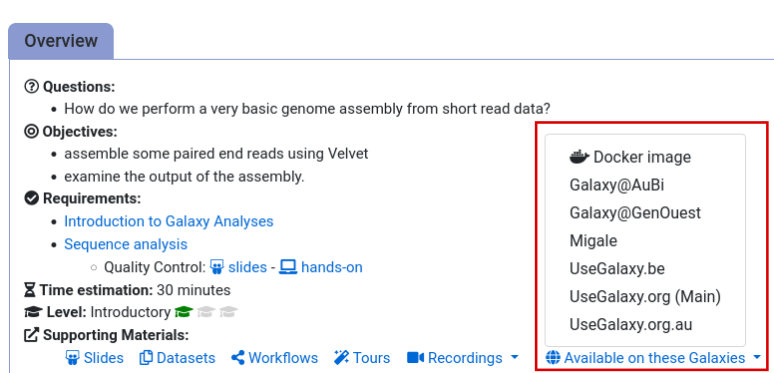
If you can’t find a tool you need for a tutorial on Galaxy, please:
- Check that you are using a compatible Galaxy server
- Navigate to the overview box at the top of the tutorial
- Find the “Supporting Materials” section
- Check “Available on these Galaxies”
- If your server is not listed here, the tutorial is not supported on your Galaxy server
- You can create an account on one of the supporting Galaxies
- Use the Tutorial mode feature
- Open your Galaxy server
- Click on the curriculum icon on the top menu, this will open the GTN inside Galaxy.
- Navigate to your tutorial
- Tool names in tutorials will be blue buttons that open the correct tool for you
- Note: this does not work for all tutorials (yet)
- Still not finding the tool?
- Ask help in Gitter.
Running into an error?
When something goes wrong in Galaxy, there are a number of things you can do to find out what it was. Error messages can help you figure out whether it was a problem with one of the settings of the tool, or with the input data, or maybe there is a bug in the tool itself and the problem should be reported. Below are the steps you can follow to troubleshoot your Galaxy errors.
- Expand the red history dataset by clicking on it.
- Sometimes you can already see an error message here
View the error message by clicking on the bug icon galaxy-bug
- Check the logs. Output (stdout) and error logs (stderr) of the tool are available:
- Expand the history item
- Click on the details icon
- Scroll down to the Job Information section to view the 2 logs:
- Tool Standard Output
- Tool Standard Error
- For more information about specific tool errors, please see the Troubleshooting section
- Submit a bug report! If you are still unsure what the problem is.
- Click on the bug icon galaxy-bug
- Write down any information you think might help solve the problem
- See this FAQ on how to write good bug reports
- Click galaxy-bug Report button
- Ask for help!
- Where?
- In the GTN Matrix Channel
- In the Galaxy Matrix Channel
- Browse the Galaxy Help Forum to see if others have encountered the same problem before (or post your question).
- When asking for help, it is useful to share a link to your history