Frequently Asked Questions
Tutorial Questions
Do I have to run the tools in the order of the tutorial?
The tools are presented in the order that a typical analysis would use. If you want to run some tools in parallel (to save time) you can do so. This workflow illustrates the analysis done in the tutorial and shows that there are multiple “paths” leading to outputs that have some steps that could be run at the same time: MultiQC, Kraken2, JBrowse and TB Variant Report.
My snippy is running for a very long time. Is this normal?
As this tutorial uses real world data some of the tools can run for quite a while. During a course we can expected longer run times as the Galaxy servers are heavily used. Typically expected runtimes are approximately:
Tool name Runtime FastQC 2 minutes MultiQC 5 minutes Trimmomatic 5 minutes kraken2 5 - 12 minutes snippy 15 - 25 minutes TB Variant Filter 2 minutes TB-Profiler 5 minutes Text transformation Less than 1 minute TB Variant Report 1 minute JBrowse 5 minutes Samtools stats (optional) 1 minute BAM Coverage plotter (optional) 1 minute
TB Variant Report crashes (with an error about KeyError: 'protein')
This is a bug present in TB Variant Report (aka tbvcfreport) version 0.1.8 and earlier. In this case it is triggered by the presence of variants in Rv3798. You only see this bug, however, if you forget to run
tb_variant_filter(TB Variant Filter). Rv3798 is a suspected transposase and any variants in this gene region would be filtered out bytb_variant_filter, so if you see this crash, make sure you have run the filter step before the TB Variant Report step.
General Questions
Can't find one of the tools for this tutorial?
To use the tools installed and available on the Galaxy server:
- At the top of the left tool panel, type in a tool name or datatype into the tool search box.
- Shorter keywords find more choices.
- Tools can also be directly browsed by category in the tool panel.
If you can’t find a tool you need for a tutorial on Galaxy, please:
- Check that you are using a compatible Galaxy server
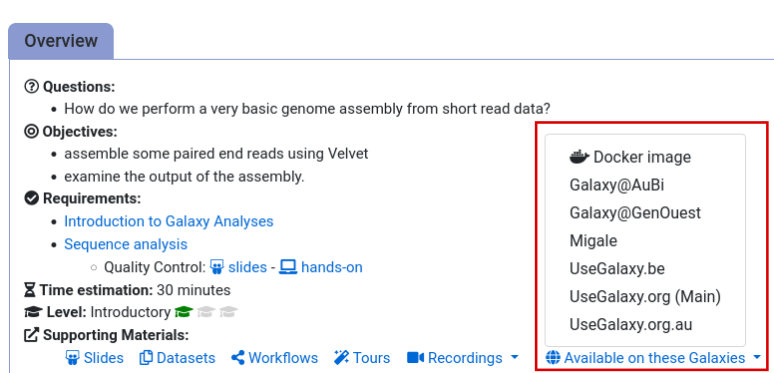
- Navigate to the overview box at the top of the tutorial
- Find the “Supporting Materials” section
- Check “Available on these Galaxies”
- If your server is not listed here, the tutorial is not supported on your Galaxy server
- You can create an account on one of the supporting Galaxies
- Use the Tutorial mode feature
- Open your Galaxy server
- Click on the curriculum icon on the top menu, this will open the GTN inside Galaxy.
- Navigate to your tutorial
- Tool names in tutorials will be blue buttons that open the correct tool for you
- Note: this does not work for all tutorials (yet)
- Still not finding the tool?
- Ask help in Gitter.
Running into an error?
When something goes wrong in Galaxy, there are a number of things you can do to find out what it was. Error messages can help you figure out whether it was a problem with one of the settings of the tool, or with the input data, or maybe there is a bug in the tool itself and the problem should be reported. Below are the steps you can follow to troubleshoot your Galaxy errors.
- Expand the red history dataset by clicking on it.
- Sometimes you can already see an error message here
View the error message by clicking on the bug icon galaxy-bug
- Check the logs. Output (stdout) and error logs (stderr) of the tool are available:
- Expand the history item
- Click on the details icon
- Scroll down to the Job Information section to view the 2 logs:
- Tool Standard Output
- Tool Standard Error
- For more information about specific tool errors, please see the Troubleshooting section
- Submit a bug report! If you are still unsure what the problem is.
- Click on the bug icon galaxy-bug
- Write down any information you think might help solve the problem
- See this FAQ on how to write good bug reports
- Click galaxy-bug Report button
- Ask for help!
- Where?
- In the GTN Matrix Channel
- In the Galaxy Matrix Channel
- Browse the Galaxy Help Forum to see if others have encountered the same problem before (or post your question).
- When asking for help, it is useful to share a link to your history