Frequently Asked Questions
Tutorial Questions
Can I use these workflows on datasets generated from our laboratory?
Yes, the workflows can be used on other datasets as well. However, you will need to consider data acquisition and sample preparation methods so that the tool parameters can be adjusted accordingly.
How does one compare metaproteomics measurements from two experimental conditions?
For comparing taxonomy composition or functional content of two conditions in metaproteomics or metatranscriptomics studies, users are recommended to use metaQuantome. GTN tutorials for metaQuantome are available in the proteomics topic.
How does one convert RAW files to MGF peak lists within Galaxy?
Galaxy has implemented msconvert tool so that RAW files from Thermo instruments can be converted into MGF or mzML formats.
I have FASTQ files from metagenomics or metatranscriptomics datasets? How can I convert them into a protein FASTA file for metaproteomics searches?
Galaxy has a tool named Sixgill that can be used to convert the nucleic acid sequences to ‘metapeptide’ sequences. There are other options available within Galaxy such as the GalaxyGraph approach and Metagenome Binning, Assembly and Annotation Workflow. Please contact us, if you need assistance.
I have a really large search database, what search strategies do you recommend for searching my mass spectrometry dataset?
Readers are encouraged to use the database sectioning approach described by Praveen Kumar et al and available within Galaxy. Readers are also encouraged to consider other approaches such as MetaNovo (not yet available in Galaxy). In absence of any database or taxonomic information about the microbiome dataset, other methods such as COMPIL 2.0 and De novo search methods can also be considered.
What other methods are available to study the functional state of the microbiome within Galaxy?
Other software such as EggNOG Mapper, MEGAN5, MetaGOmics, MetaProteomeAnalyzer (MPA) and ProPHAnE also generate functional outputs.
What software tools are available to determine taxonomic composition from mass spectrometry data?
Within the Galaxy framework we recommend the use of Unipept software that uses NCBI taxonomy and UniProt databases to detect unique peptides for taxonomy. Other software tools such as MetaTryp 2.0 (PMID: 32897080) can also be used to determine the taxonomic composition of the metaproteomics datasets.
Which search algorithms are recommended for searching the metaproteomics data?
SearchGUI supports search using nine search algorithms (X! Tandem. MS-GF+. OMSSA, Comet, Tide, MyriMatch, MS_Amanda, DirecTag and Novor). For this tutorial, we have used the first two search algorithms in the list. In our hands, the first four search algorithms have given us the most optimal results.
Which version of SearchGUI and PeptideShaker shall I use for this tutorial?
We highly recommend the usage of SearchGUI Galaxy version 3.3.10.1 and PeptideShaker version Galaxy Version 1.16.36.3. The newer versions of SearchGUI and PeptideShaker have not yet been tested for this workflow.
Why do we do dimension reduction and then clustering? Why not just cluster on the actual data?
Within the Galaxy framework we recommend the use of Unipept software that uses UniProt databases and annotation to detect proteins (EC terms) and functional groups such as GO Ontology and InterPro terms. Other software tools such as EggNOG Mapper are also available within the Galaxy platform. Other software such as MEGAN5, MetaGOmics, MetaProteomeAnalyzer (MPA), ProPHAnE also generate functional outputs.
General Questions
Can't find one of the tools for this tutorial?
To use the tools installed and available on the Galaxy server:
- At the top of the left tool panel, type in a tool name or datatype into the tool search box.
- Shorter keywords find more choices.
- Tools can also be directly browsed by category in the tool panel.
If you can’t find a tool you need for a tutorial on Galaxy, please:
- Check that you are using a compatible Galaxy server
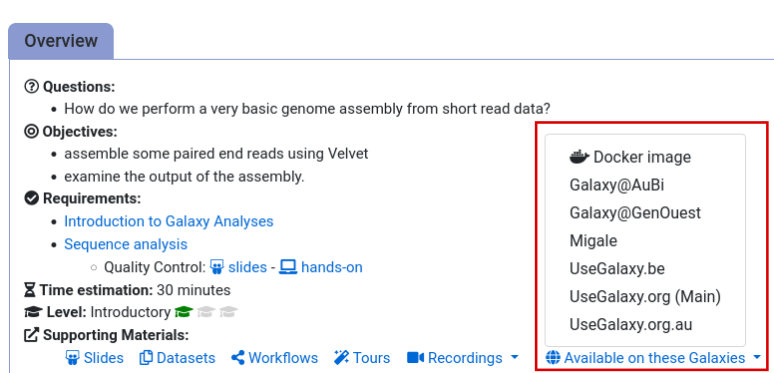
- Navigate to the overview box at the top of the tutorial
- Find the “Supporting Materials” section
- Check “Available on these Galaxies”
- If your server is not listed here, the tutorial is not supported on your Galaxy server
- You can create an account on one of the supporting Galaxies
- Use the Tutorial mode feature
- Open your Galaxy server
- Click on the curriculum icon on the top menu, this will open the GTN inside Galaxy.
- Navigate to your tutorial
- Tool names in tutorials will be blue buttons that open the correct tool for you
- Note: this does not work for all tutorials (yet)
- Still not finding the tool?
- Ask help in Gitter.
Running into an error?
When something goes wrong in Galaxy, there are a number of things you can do to find out what it was. Error messages can help you figure out whether it was a problem with one of the settings of the tool, or with the input data, or maybe there is a bug in the tool itself and the problem should be reported. Below are the steps you can follow to troubleshoot your Galaxy errors.
- Expand the red history dataset by clicking on it.
- Sometimes you can already see an error message here
View the error message by clicking on the bug icon galaxy-bug
- Check the logs. Output (stdout) and error logs (stderr) of the tool are available:
- Expand the history item
- Click on the details icon
- Scroll down to the Job Information section to view the 2 logs:
- Tool Standard Output
- Tool Standard Error
- For more information about specific tool errors, please see the Troubleshooting section
- Submit a bug report! If you are still unsure what the problem is.
- Click on the bug icon galaxy-bug
- Write down any information you think might help solve the problem
- See this FAQ on how to write good bug reports
- Click galaxy-bug Report button
- Ask for help!
- Where?
- In the GTN Matrix Channel
- In the Galaxy Matrix Channel
- Browse the Galaxy Help Forum to see if others have encountered the same problem before (or post your question).
- When asking for help, it is useful to share a link to your history