Frequently Asked Questions
Tutorial Questions
After sequencing with MinKNOW software, we get many fastq files, do these files need to be combined into one file before uploading or is it possible to upload them all at once?
After sequencing with MinKNOW software, it is a good approach to combine the files from the same run before processing them. You could create a collection per run with all fastq files and then use the collection operation to concatenate all files in a collection.
Can we also use this workflow on Illumina raw reads?
Yes, some tools would need to be changed or removed:
- For the Preprocessing workflow, plotting with Nanoplot shall be removed and keep only FastQC, MultiQC and Fastp.
- For the mapping in the SNP based pathogen detection workflow, instead of Minimap2, Bowtie can be used.
Can we use snippy pipeline instead for the phylogenetic analysis?
On principle yes. We did not try yet. Snippy is available in Galaxy
Do the pipelines work with both isolates and direct from raw meat? or only isolate?
The workflow can work with both isolates and raw meat. The workflow is designed to remove hosts before detecting any pathogen, so both isolates and raw meat samples are pre-processed equaliy before the analysis starts.
For preprocessing part with host removal: Where do you find the abbreviations for each host species available (e.g. bos is cow, homo is human..)?
The abbreviation (i.e. the genus) is the first word in the list of possible hosts. The names are the scientific names for species, which would be shown on the taxonomy tree if you would look up the common name (i.e. bovine) on Wikipedia.
From where can I import other genomes?
In this tutorial, we used kalamari DB with the full list of possible host sequences that can be removed. Reads are either tagged to map one of those species or are left unassigned. If the task at hand in the real world cannot be covered by those, you can also try another DB for Kraken2 that includes your species (or maybe retain unmapped reads from a read aligner such as Bowtie2, Minimap2…).
Is there a way to filter on the Kalimari database?
To filter the Kalamari database, e.g. leaving out milk bacteria only to detect spoilers or contaminants, but the Kalimera list contains a lot more than that, you can:
- Look at a publication etc. to find a list of bacteria to remove.
- Change the regex
^.*Gallus|Homo|Bos.*$to^.*Gallus|Homo|Bos|Bacterium1|Bacterium2...|BacteriumN.*$Milk pathogens are somewhat known, Salmonella, Escherichia… It might be easier to retain reads only mapping to pathogens instead
Isn't it awkward to find so many humans sequences there, since we filter for them before?
We see a lot that Kraken tends to assign many reads to human, despite they do not map to human genome. Due to resemblance between organisms and the limited species coverage of Kraken databases sometimes does happen that reads corresponding to higher organisms get mapped to humans. It was a very severe problem for the standard databases, because yeast genes were mis-assigned to human.
What does `^.*Gallus|Homo|Bos.*$` mean?
^.*Gallus|Homo|Bos.*$is a regular expression that matches a string containing the words Gallus OR Homo OR Bos.
General Questions
Can't find one of the tools for this tutorial?
To use the tools installed and available on the Galaxy server:
- At the top of the left tool panel, type in a tool name or datatype into the tool search box.
- Shorter keywords find more choices.
- Tools can also be directly browsed by category in the tool panel.
If you can’t find a tool you need for a tutorial on Galaxy, please:
- Check that you are using a compatible Galaxy server
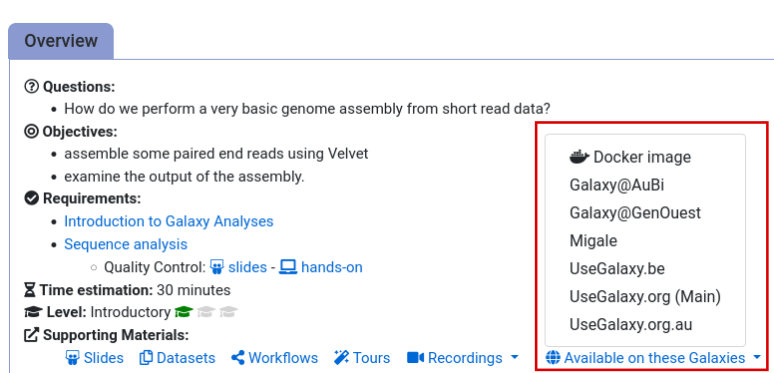
- Navigate to the overview box at the top of the tutorial
- Find the “Supporting Materials” section
- Check “Available on these Galaxies”
- If your server is not listed here, the tutorial is not supported on your Galaxy server
- You can create an account on one of the supporting Galaxies
- Use the Tutorial mode feature
- Open your Galaxy server
- Click on the curriculum icon on the top menu, this will open the GTN inside Galaxy.
- Navigate to your tutorial
- Tool names in tutorials will be blue buttons that open the correct tool for you
- Note: this does not work for all tutorials (yet)
- Still not finding the tool?
- Ask help in Gitter.
Running into an error?
When something goes wrong in Galaxy, there are a number of things you can do to find out what it was. Error messages can help you figure out whether it was a problem with one of the settings of the tool, or with the input data, or maybe there is a bug in the tool itself and the problem should be reported. Below are the steps you can follow to troubleshoot your Galaxy errors.
- Expand the red history dataset by clicking on it.
- Sometimes you can already see an error message here
View the error message by clicking on the bug icon galaxy-bug
- Check the logs. Output (stdout) and error logs (stderr) of the tool are available:
- Expand the history item
- Click on the details icon
- Scroll down to the Job Information section to view the 2 logs:
- Tool Standard Output
- Tool Standard Error
- For more information about specific tool errors, please see the Troubleshooting section
- Submit a bug report! If you are still unsure what the problem is.
- Click on the bug icon galaxy-bug
- Write down any information you think might help solve the problem
- See this FAQ on how to write good bug reports
- Click galaxy-bug Report button
- Ask for help!
- Where?
- In the GTN Matrix Channel
- In the Galaxy Matrix Channel
- Browse the Galaxy Help Forum to see if others have encountered the same problem before (or post your question).
- When asking for help, it is useful to share a link to your history