EWAS Epigenome-Wide Association Studies Introduction
Contributors
What is the topic?
Epigenome-wide association studies (EWAS) analyse genome-wide activity of epigenetic marks in cohorts of different individuals to find associations between epigenetic variation and phenotype.
Overview
- Applications of EWAS
- Infinium Human Methylation BeadChip
- The Analysis
-
Integrative Approach
Applications
- Population Studies
- EWAS studies are the solution to explore gene-environment interactions on big scale
- Clinical
- Genetic epidemiology is widely used in many progressive and incurable diseases i.e. cancers, autoimmune and neurological disorders
.image-75[ 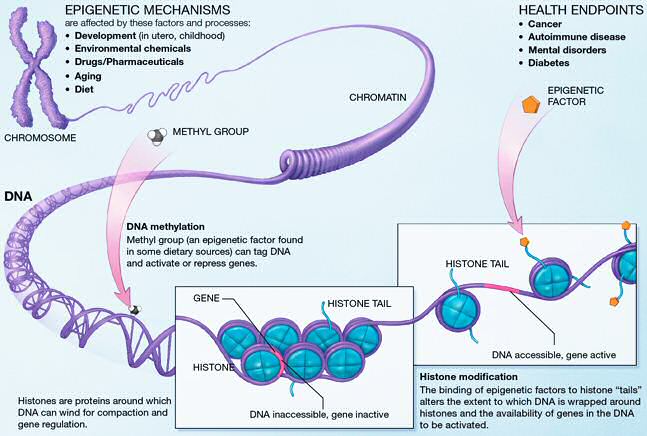 ]
]
National Institutes of Health, 2005
Infinium Human Methylation BeadChip
One of the most common techniques used in EWAS studies is the Infinium Human Methylation BeadChip array quantifying the DNA methylation level, providing high accuracy with low input DNA requirements.
.image-80[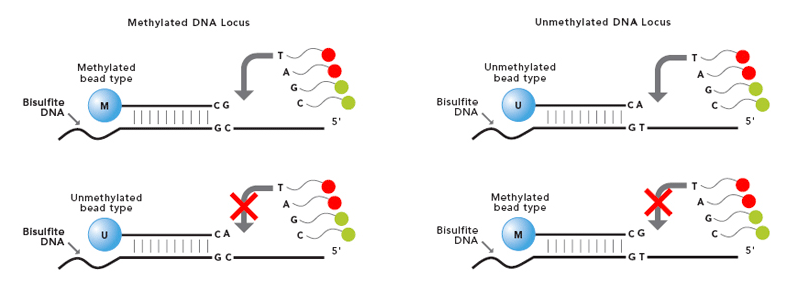 ]
]
The Infinium Methylation Assay uses two different bead types to detect changes in DNA methylation levels. In the figure we can see M - methylated and U - unmethylated bead types. Depending on the probe design, the bead signals are reported in different colors green or red.
The Analysis
.image-90[ ]
]
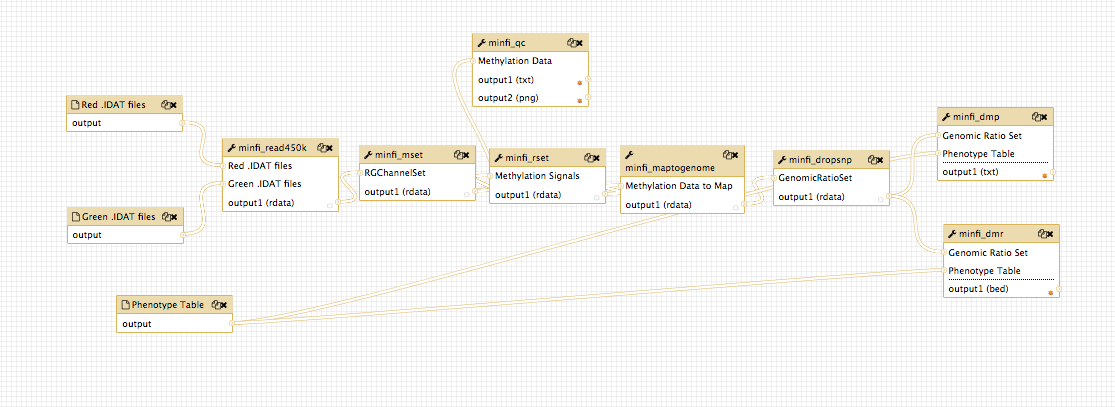
The analysis combines 6 main steps:
- Raw intensity data loading (.idat files)
- Preprocessing and optional normalization
- Quality assessment and control step
- Differentially methylated positions (DMP) and regions (DMR) finding with respect to a phenotype covariate
- Functional annotation and graphical representation
</small>
.image-30[
 ]
—
]
—
Integrative Approach
Methylation patterns obtained from Infinium Human Methylation BeadChip data are compatibile with other platforms (gene expression, microRNA profiling) and thus become one of the most comprehensive solution to invastigate epigenome.
Summary
Epigenetic aberrations which involve DNA modifications give researchers an interest to identify novel non-genetic factors responsible for complex human phenotypes such as height, weight, and disease. To identify methylation changes researchers need to perform complicated and time consuming computational analysis.
Here, the Infinium Human Methylation BeadChip pipeline becomes a solution for this inconvenience and provides a simplified downstream analysis including preprocessing, quality evaluation and differentially methylated regions detection in one complex set of tools developed and published under the Galaxy platform. Ready to be integrated with existing resources and reused in multiple ways.
Thank you!
This material is the result of a collaborative work. Thanks to the Galaxy Training Network and all the contributors! Tutorial Content is licensed under
Creative Commons Attribution 4.0 International License.
Tutorial Content is licensed under
Creative Commons Attribution 4.0 International License.